Meesmann Corneal Dystrophy
All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.
Meesmann Corneal Dystrophy is a rare genetic autosomal dominant corneal disease characterized by fragility of the anterior corneal epithelium.
Disease Entity
- Meesmann Corneal Dystrophy ICD-10: H18.5
- Disease Synonyms : MECD, Juvenile Hereditary Epithelial Dystrophy, Meesmann-Wilke syndrome
Disease

{kind=link}

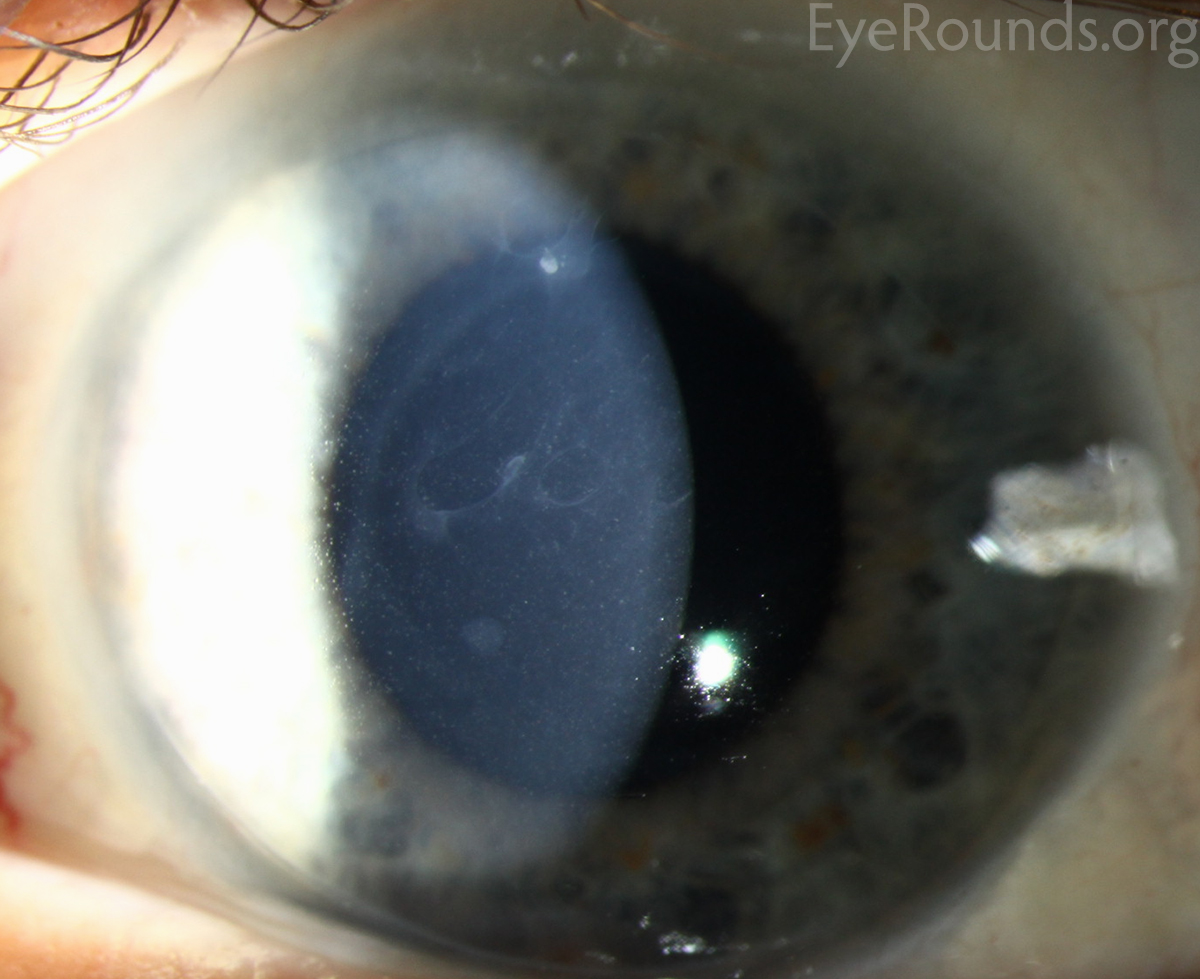
Meesmann Corneal Dystrophy (MECD) is a rare hereditary type of superficial corneal dystrophy that follows an autosomal dominant pattern of inheritance. It is characterized by the bilaterally symmetric development of intraepithelial microcysts that cause fragility of the anterior corneal epithelium. Unlike most other corneal dystrophies, pathology in MECD manifests early in life and epithelial cysts may present already in the first years of life. The onset of symptoms during infancy or early childhood does not become noticeable or problematic until adulthood. As the disorder slowly progresses, the number of tiny epithelial vesicles usually increases with age[3] involving central visual axis and mid-periphery of the cornea[4] and can extend out to the limbus. The bubble-like blebs tend to be denser in the interpalpebral area and can be best visualized with retroillumination[5] Most patients remain asymptomatic or may experience minor symptoms and minimal vision disturbances for years, while some suffer from recurrent episodes of corneal erosions resulting in lacrimation, photophobia, and deterioration in visual acuity, accompanied by irregular astigmatism and subepithelial scarring in some severe cases[3], particularly in elderly patients. Vision is seldom affected to a significant degree.
History
The disorder is named after German ophthalmologist Alois Meesmann (1888-1969)[6][7], who was the first along with F. Wilke to describe the histopathological features of this form of corneal dystrophy in 1939. It is often also referred to as the "Meesmann-Wilke syndrome", after the joint contribution of Meesmann and Wilke, based on the observation of 3 affected families in Schleswig-Holstein, Germany.[7][8] Further research was later contributed by Stocker and Holt in 1954 through 1955, who discovered a variant of Meesmann corneal dystrophy called "Stocker-Holt Dystrophy"[9], after identifying a recessive form in 20 members of a large family living in North Carolina, USA. The family was of Moravian descent and had emigrated from Saxony in 1741.[10]
Etiology
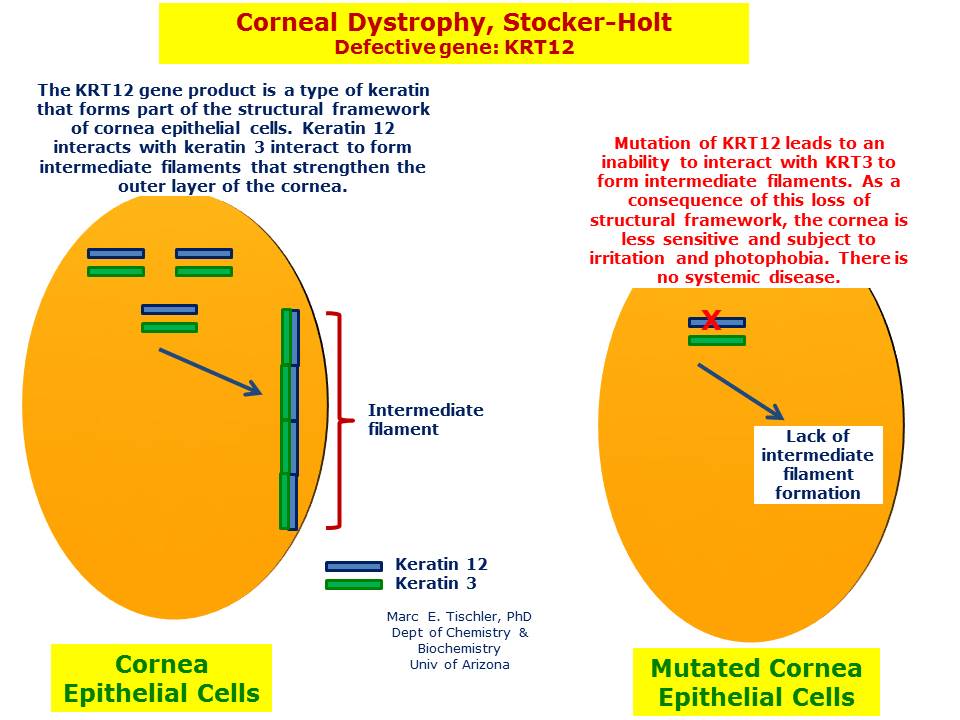
MECD is a genetically heterogeneous autosomal dominant disorder resulting from mutations in either one of the pair of genes KRT3 (12q13.13) or KRT12 (17q11-q1), that are responsible for encoding the two cornea-specific cytokeratin units in the epithelium.[11] There are two phenotypes described as Meesmann corneal dystrophy 1 (MECD1) and Meesmann corneal dystrophy 2 (MECD2), that are caused by mutations in genes KRT3 and KRT12 respectively. A heterozygous mutation in either of these genes leads to a single dystrophy phenotype.[12][13] The two affected protein units normally interact to form the structural framework of the epithelium; however, the defective keratin lacks the strength of the normal corneal structure and is more susceptible to damage. Mutations in either the KRT12 or KRT3 gene weaken this framework, leading to fragility of the corneal epithelium and the development of bubble-like vesicles, specific to the disorder. The cysts contain clumps of abnormal keratin proteins and other cellular debris and their rupture causes ocular irritation.[14]
{kind=link}
Epidemiology
The prevalence of MECD is unknown, as an official registry of affected cases does not exist.[11] It was first described in a large, multi-generational German family with more than 100 affected members and since then, the condition has been reported in individuals and families worldwide. Numerous cases have been identified in Denmark, Germany, the Netherlands, Northern Ireland, Switzerland, the USA, Japan, Saudi Arabia, Taiwan, and Poland.[10][15]
General Pathology

The primary pathology of MECD lies in the corneal epithelium and its basement membrane. Histopathologically, it is described by the development of clusters of small, clear intraepithelial cysts diffusely distributed at different levels within the corneal epithelium, which is irregular in thickness.[11] The transparent microcysts are roughly the same size and are mostly found in the basal epithelial cells. They contain degenerated epithelial cells and cellular debris that are periodic acid-Schiff (PAS) positive[4] and diastase- and neuraminidase- resistant.[11] Degenerated cellular debris within the intraepithelial microcysts manifests autofluorescence in ultraviolet (UV) light and it stains with the Hale colloidal iron technique for negatively charged substances such as glycosaminoglycans (GAGs). Other distinct pathological findings include the presence of electron-dense filamentary and granular material, the pathognomonic so-called peculiar substance, in the epithelial cells, and vacuolated, homogenous substance in the epithelial cysts (most commonly) and epithelial cells.[4] They may coalesce to form refractile lines and become more opaque.[17] The epithelium adjacent to the cysts remains clear.[5] Light microscopy shows the epithelial basement membrane to appear coarse, variably abnormally thickened, and multilaminar. Bowman layer and the corneal stroma are spared and remain intact.
Diagnosis
History
Although the presence of intraepithelial cysts becomes evident in the first decade of life,[4] the vast majority of patients tend to remain asymptomatic or experience mild symptoms that often go unnoticed until late adolescence or adulthood. Affected individuals are therefore often diagnosed incidentally during a routine ophthalmic examination.[18] Overall, the disorder does not cause much discomfort or significant vision disturbances[19] and symptoms are usually mild and include recurrent irritation and decrease in visual acuity. The first presented symptom is ocular irritation linked to recurrent corneal erosions such as photophobia, pain, foreign body sensation, and excessive tearing, particularly upon lid-opening during sleep or upon awaking.[20] The second symptom that follows is blurred vision as a result of corneal clouding.[20]
Signs
- Myriads of intraepithelial microcysts most prominently in the interpalpebral zone
- Round-to-oval shaped punctate opacities in the central corneal epithelium[11]
- Recurrent corneal erosions
- Reduced corneal sensitivity
- Smooth corneal surface
Symptoms
- Decreased visual acuity (usually minor)
- Foreign body sensation
- Photophobia
- Lacrimation
- Temporary episodes of blurred vision
- Eye stinging
- Blepharospasm
Clinical Diagnosis
Slit-lamp examination in direct and indirect illumination, retroillumination, fluorescein staining, preferably with dilated pupil.
Diagnostic Procedures

- Slit-lamp examination in direct illumination: the lesions appear as a variety of different patterns (random, whirled, sectorial, interpalpebral or unilateral and sparing the perilimbal region) of multiple tiny grey dot-like opacities and tiny vesicles in the corneal epithelium, that tend to concentrate in the interpalpebral zone.[10]Fluorescein usually fails to stain the microcysts, as they rarely open to the corneal surface.[11]
- Slit-lamp examination in indirect illumination: the grey opacities appear as multiple solitary, round, and transparent cysts, rarely producing refractile lines due to a coalescence of several cysts[10], while the intervening cornea remains intact or slightly hazy.
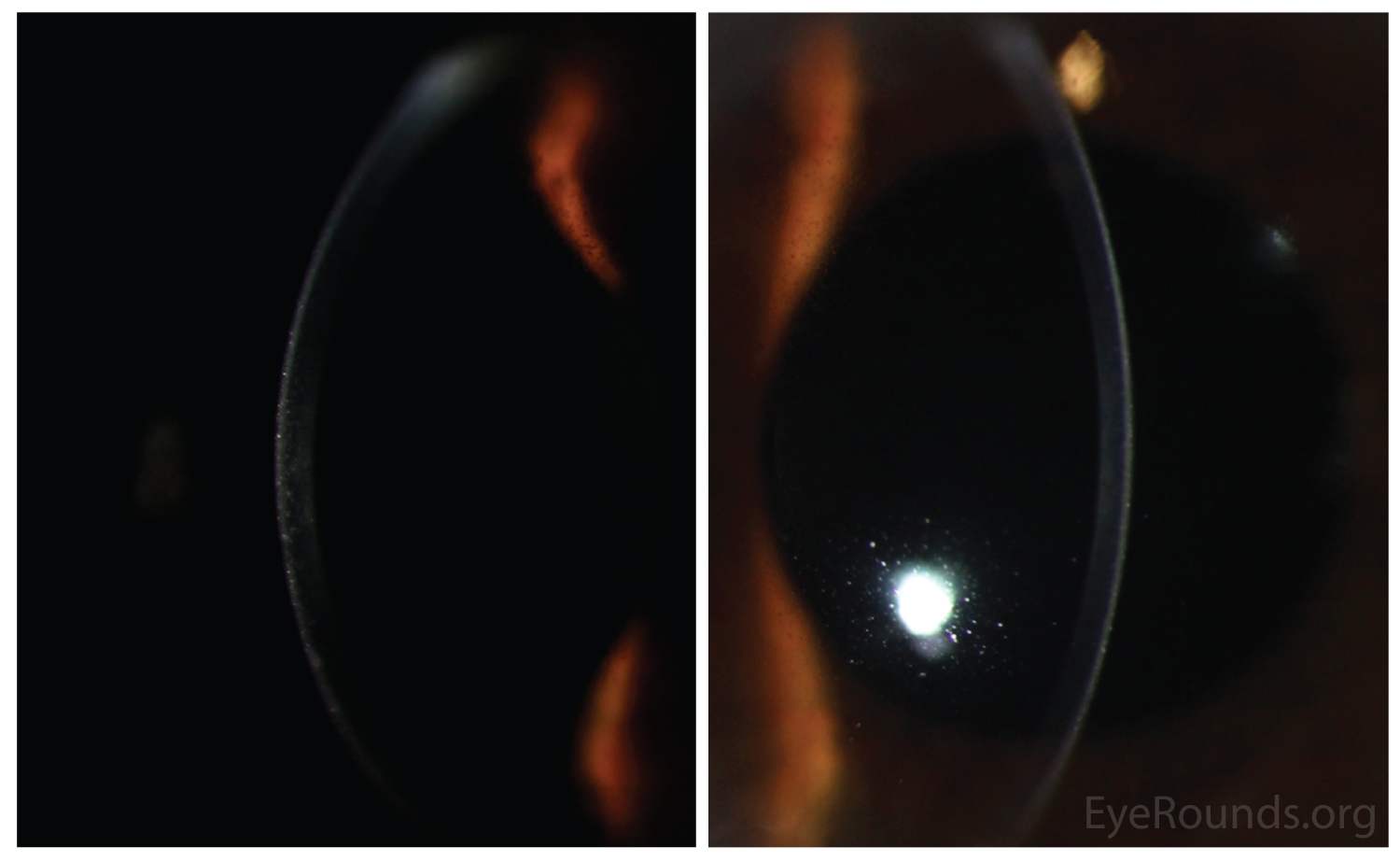
- Retroillumination: the intraepithelial microcysts appear as refractile transparent dew drops[11] and the vesicles present isolated.[22] The characteristic bleb pattern of Meesmann corneal dystrophy can be best seen upon retroillumination.[23]
- Corneal elevation topography: marked irregularity of Placido rings
- In vivo confocal microscopy (IVCM): the microcysts present as hyporeflective, round-oval, or teardrop areas in the basal epithelial cell layer. The peculiar substance appears as reflective spots.[24]

{kind=link}
- Slit-Lamp Biomicroscopy and Photography: documentation of the biomicroscopic examination with photos is useful as the disease progresses.
Differential diagnosis
Suspected cases of MECD need to be differentiated from other disorders of the corneal epithelium, such as:
- Vapor spray keratitis
- Epithelial basement membrane dystrophy or Cogan's microcystic corneal dystrophy
- Reis-Bücklers corneal dystrophy.
- Lisch epithelial corneal dystrophy (LECD)
MECD and LECD appear to present clinical similarities, and the two disease entities need to be distinguished from each other. This can be achieved by studying the different patterns of inheritance, i.e. X-linked recessive versus autosomal dominant.[11]
Management
General treatment
Meesmann dystrophy is a genetic disorder that persists throughout life.[11]MECD usually remains an entirely asymptomatic condition that requires no treatment.[12] For patients who experience minor intermittent symptoms of ocular irritation, a conservative therapeutic approach largely aimed at symptoms is preferred. In rare cases of severe symptomatology or recurrent episodes of corneal erosions, surgical interventions may be required to prolong the reoccurrence of the disorder, and also lessen its severity.[12]
Medical therapy
Mild to moderate symptoms and MECD-associated chronic eye dryness can be treated by using lubricating eye drops during the daytime and gel/ointment at night, but the majority of patients do not usually require further treatment.[12] Some physicians advocate the use of topical steroids to increase the stability of the epithelial basement membrane, while others prefer hypertonic saline, especially in gel or ointment form at night, which causes dehydration of the epithelium and promotes its attachment to the underlying layers. [20] Patching, either conventional or using a bandage soft contact lens, can be beneficial as it minimizes the mechanical effect of lid movement on the irregular corneal epithelium.[20][26]
Surgery
A more severe phenotype of MECD, described by recurrent corneal erosions and secondary scarring, can complicate the clinical presentation of MECD and conservative therapy often proves to be unsuccessful. Surgical treatment options aim at the removal of the pathological corneal epithelium and include superficial corneal debridement, superficial keratectomy with or without mitomycin C, phototherapeutic keratectomy (PTK), lamellar keratoplasty, and penetrating keratoplasty. Although the dystrophy frequently recurs after any of these treatments, in some cases recurrence may be less severe, delayed, or not occur.
Superficial Mechanical Debridement of the epithelium can be useful in the management of RCE, but is usually followed by reappearance of symptoms.[27]
Superficial Keratectomy is a safe technique for managing corneal erosions and although it may be curative, it is rarely warranted.
Phototherapeutic Keratectomy using a 193- nm excimer laser is the method of choice nowadays, replacing manual lamellar keratoplasty as the former treatment of choice.[20] PTK can be repeated several times for the treatment of recurrent corneal erosions, thus postponing corneal transplantation (lamellar or even penetrating) for a long time. This treatment achieves the removal of superficial corneal opacities, smoothing of the corneal surface, as well as correcting irregular astigmatism, and finally promotes the adherence of the epithelium to the underlying layers.[10]
Lamellar and Penetrating Keratoplasty are seldom indicated for the treatment of MECD.
Rarely, a more severe phenotype with corneal erosions and significant scarring accompanied by a marked decrease in visual acuity may require treatment by keratoplasty or corneal grafting.[28] Although epithelium damage is likely to recur in grafts, the recurrent disease is often less severe.[23] It is important to postpone grafting as long as possible because most patients require regrafting due to recurrences. [10]
Surgical follow up
Surgical procedures that aim to remove the abnormal corneal epithelium are not curative, as the dystrophy recurs in the regenerated epithelium, and therefore provide only temporary symptomatic relief. All surgical treatment options have a high risk of recurrence.
Investigative Therapies
Limbal stem cell allo-transplantation may be an alternative to replace the affected epithelial stem cells, while inhibition of the abnormal gene using specific siRNA’s to silence the mutant allele has been evaluated in vitro and is a promising potential treatment strategy.[28]
Prognosis
The prognosis for these patients remains mostly favorable. In rare cases, vision can be permanently affected as a result of secondary scarring with vesicular rupture.
References
- ↑ Contributor : Christopher Kirkpatrick. Eye Rounds Org. University of Iowa
- ↑ Klintworth Orphanet Journal of Rare Diseases 2009 4:7 doi:10.1186/1750-1172-4-7
- ↑ 3.0 3.1 Goldberg A, Schlötzer-Schrehardt U, Seiler T. Unilateral Meesmann's dystrophy. Int Ophthalmol. 1997;21(3):117‐120. doi:10.1023/a:1026456232212
- ↑ 4.0 4.1 4.2 4.3 Schmidt-Erfurth U, Kohnen T. Juvenile Epithelial Dystrophy (Meesmann Dystrophy. In: Encyclopedia of Ophthalmology. Berlin: Springer; 2018:977-988.
- ↑ 5.0 5.1 Prajna NV, Peyman GA, Hamada S, Maqsood S. Ch 69 : Corneal Disorders in Children. In: Peyman's Principles & Practice of Ophthalmology. Vol 1. 2nd ed. New Delhi: Jaypee Brothers; 2019:1183.
- ↑ synd/3139 at Who Named It?
- ↑ 7.0 7.1 Meesmann. Klinische und anatomische Untersuchungen über eine bisher unbekannte, dominant vererbte Dystrophia epithelialis corneae. Bericht der Deutschen ophthalmologischen Gesellschaft, Heidelberg, 1938, 52: 154-158.
- ↑ A. Meesmann, F. Wilke. Klinische und anatomische Untersuchungen über eine bisher unbekannte, dominant vererbte Epithel Dystrophie der Horn haut. Klinische Monatsblätter für Augenheilkunde, Stuttgart, 1939, 103: 361-391.
- ↑ Online Mendelian Inheritance in Man (OMIM) 122100
- ↑ 10.0 10.1 10.2 10.3 10.4 10.5 Lisch W., Janecke A., Seitz B. (2009) Corneal Dystrophy, Meesmann. In: Lang F. (eds) Encyclopedia of Molecular Mechanisms of Disease. Springer, Berlin, Heidelberg
- ↑ 11.0 11.1 11.2 11.3 11.4 11.5 11.6 11.7 11.8 Klintworth, G.K. Corneal dystrophies. Orphanet J Rare Dis 4, 7 (2009). https://doi.org/10.1186/1750-1172-4-7
- ↑ 12.0 12.1 12.2 12.3 Greiner, Jack V.; Lindsay, Michael E.; Kenyon, Kenneth R.; Herman, John P.; Reddy, Chaitanya V. (December 2017). "Meesmann epithelial corneal dystrophy: recurrence following photorefractive keratectomy". Canadian Journal of Ophthalmology. 52 (6): e211–e213. doi:10.1016/j.jcjo.2017.05.009. ISSN 0008-4182.
- ↑ Allen, Edwin H.A.; Courtney, David G.; Atkinson, Sarah D.; Moore, Johnny E.; Mairs, Laura; Poulsen, Ebbe Toftgaard; Schiroli, Davide; Maurizi, Eleonora; Cole, Christian; Hickerson, Robyn P.; James, John (2016-01-11). "Keratin 12 missense mutation induces the unfolded protein response and apoptosis in Meesmann epithelial corneal dystrophy". Human Molecular Genetics. 25 (6): 1176–1191. doi:10.1093/hmg/ddw001. ISSN 0964-6906
- ↑ Meesmann corneal dystrophy - Genetics Home Reference - NIH. U.S. National Library of Medicine. https://ghr.nlm.nih.gov/condition/meesmann-corneal-dystrophy. Published May 26, 2020. Accessed June 1, 2020.
- ↑ Szaflik JP, Ołdak M, Maksym RB, et al. Genetics of Meesmann corneal dystrophy: a novel mutation in the keratin 3 gene in an asymptomatic family suggests genotype-phenotype correlation. Mol Vis. 2008;14:1713‐1718. Published 2008 Sep 15.
- ↑ Klintworth, G.K. Corneal dystrophies. Orphanet J Rare Dis 4, 7 (2009). https://doi.org/10.1186/1750-1172-4-7
- ↑ Allen, Meesmann, Irvine AD. Corneal Dystrophy, Meesmann. Hereditary Ocular Disease. https://disorders.eyes.arizona.edu/disorders/corneal-dystrophy-meesmann. Accessed June 1, 2020.
- ↑ Cremona, Federico A; Ghosheh, Faris R; Laibson, Peter R; Rapuano, Christopher J; Cohen, Elisabeth J (April 2008). "Meesmann Corneal Dystrophy Associated With Epithelial Basement Membrane and Posterior Polymorphous Corneal Dystrophies". Cornea. 27 (3): 374–377. doi:10.1097/ico.0b013e31815c18fa. ISSN 0277-3740.
- ↑ Burns RP. Meesmann’s corneal dystrophy. Trans Am Ophthalmol Soc. 1968;66:530–535.
- ↑ 20.0 20.1 20.2 20.3 20.4 Gault JA, Vander JF, Hannush SB, Riveroll-Hannush L. Ch 12 : Corneal Dystrophies . In: Ophthalmology Secrets in Color. 4th ed. Philadelphia, PA: Elsevier; 2016:115-116.
- ↑ Klintworth Orphanet Journal of Rare Diseases 2009 4:7 doi:10.1186/1750-1172-4-7
- ↑ Kivelä T., Messmer E.M., Rymgayłło-Jankowska B. (2015) Cornea. In: Heegaard S., Grossniklaus H. (eds) Eye Pathology. Springer, Berlin, Heidelberg
- ↑ 23.0 23.1 Foster CS, Azar DT, Dohlman CH, Charukamnoetkanok P, Tuli S, Azar DIT. Ch 45 : Anterior corneal dystrophies. In: Smolin and Thoft's The Cornea: Scientific Foundations and Clinical Practice. Philadelphia: Wolters Kluwer; 2015.
- ↑ Lisch W, Vemuganti GK, Rathib VM, Murthy SI. Histological Landmarks in Corneal Dystrophy: Pathology of Corneal Dystrophies. In: Corneal Dystrophies: 13 Tables. Vol 48. Basel: Karger, S; 2011:27.
- ↑ Klintworth Orphanet Journal of Rare Diseases 2009 4:7 doi:10.1186/1750-1172-4-7
- ↑ Bourne W. Soft contact lens wear decreases epithelial microcysts in Meesmann’s corneal dystrophy. Trans Am Ophthalmol Soc 1986;84: 170–81.
- ↑ Tasman W, Jaeger EA. Ch 1. External Disease : Epithelial Corneal Dystrophies . In: The Wills Eye Hospital Atlas of Clinical Ophthalmology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:43.
- ↑ 28.0 28.1 Liao, H., Irvine, A. D., Macewen, C. J., Weed, K. H., Porter, L., Corden, L. D., Gibson, A. B., Moore, J. E., Smith, F. J., McLean, W. H., Moore, C. B. Development of allele-specific therapeutic siRNA in Meesmann epithelial corneal dystrophy. PLoS One 6: e28582, 2011.