All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.
Disease Entity
Crystalline retinopathies are a heterogenous group of disorders associated with crystal deposits in any layer and/or region of the retina.[1][2][3][4] The crystals can be composed of multiple different substances, and the causes can be categorized into genetic, toxic, degenerative, idiopathic, and iatrogenic. Establishing the diagnosis is based upon history, specific exam findings and multimodal imaging features unique to each disease.[3]
Due to the heterogeneous nature of causes, this article will distinctly discuss each of the causes, risk factors, general pathology, pathophysiology, primary prevention, diagnosis, and management when possible.
Etiologies


Genetic
- Bietti crystalline dystrophy (BCD) is an inherited (autosomal recessive) tapetoretinal and marginal corneal dystrophy caused by a mutated CYP4V2 gene on chromosome 4q35 which encodes for specific cytochrome P450 involved in the oxidation of ocular fatty acids, fibroblasts and circulating lymphocytes.[6][7][8] Though the exact mechanism is unknown, the retinal pigment epithelium (RPE) and choriocapillaris atrophy lead to secondary photoreceptor degeneration from crystal accumulation. Risk factors include Italian or East Asian descent.[9] Vision loss that progresses rapidly can be due to cystoid macular edema (CME), choroidal neovascularization (CME), and macular hole (MH) formation.[3]
- Diagnosis is made when the following triad of symptoms are present: progressive nyctalopia, reduced visual acuity, and visual field loss in the third decade of life.[6][10] Slit-lamp exam can reveal crystals in the perilimbal anterior corneal stroma.[11] On dilated fundus exam (DFE), numerous prominent small, glistening yellow-white crystals are seen scattered throughout the posterior pole extending to the midperiphery. These crystalline deposits may tend to diminish with advanced disease. The yellow-white intraretinal crystals are found at the level of RPE-choriocapillaris complex and lead to RPE pigment clumping and choriocapillaris atrophy. The classic triad of symptoms is also appreciated in diseases on spectrum of retinitis pigmentosa and should be considered in the differential diagnosis.[12] With few ocular diseases sharing common findings of the typical crystalline deposits in the cornea and retina, diagnosis can be made with high degree of certainty from clinical exam alone.[8] There are three stages described by Yuzawa et al[13]:
- Stage 1 BCD: crystalline deposits in posterior pole and midperiphery with mild macular RPE atrophy
- Stage 2 BCD: progressive RPE atrophy extending beyond the posterior pole with associated chorioretinal atrophy with crystals diminishing in the posterior pole but remaining mid-periphery
- Stage 3 BCD: diffuse RPE and choriocapillaris atrophy with few to no crystals
- Diagnostic imaging to evaluate for BCD includes fluorescein angiography (FA), indocyanine green angiography (ICGA), fundus autofluorescence (FAF), spectral domain optical coherence tomography (SD-OCT), near-infrared reflectance (NIR) imaging, and electroretinogram (ERG). Intraretinal crystals do not exhibit hyper- or hypo-fluorescence or hyper- or hypo-cyanescence and have no masking effect on FA or ICGA.[14] Areas of RPE atrophy correspond to hyperfluorescent window defects on FA. As the disease progresses to chorioretinal atrophy, the FA shows nummular hypofluorescent lesions.[6] ICGA findings vary with course of the disease but all stages have delayed choroidal filling that progresses with severity of disease and derangement of inner choroidal circulation with late lobular hypocyanescence. ICGA is superior to FA in delineating the full extent of RPE-choriocapillaris atrophy and choroidal vascular compromise (decreased choroidal vessel volume and sclerosed vasculature).[14][15] Though FAF does not show crystalline deposits, it does illustrate granular hyperautofluorescence in distressed RPE cells that progress to areas of hypoautofluorescence as RPE cells atrophy. En-face near-infrared image highlights crystals well.[16] Full-field ERG; depending on the stage of disease, can be normal, reduced, or extinguished. There is progressive depression of the ERG amplitudes with rod and cone waveforms diminishing often in parallel.[17] Waveforms can also demonstrate cone-rod pattern of dysfunction which is more common than the rod-cone pattern.[18] SD-OCT show hyperreflective retinal dot in all the layers of the retina with majority found in the RPE-Bruch membrane complex.[6] They are not found in the choroid.[19] Other common findings are outer retinal tubulations (ORTs) which are observed as circular hyperreflective-outlined structures in the outer nuclear layer that cross-sections of spherical structures in areas where Muller cells react to nonviable photoreceptors.[20][21] SD-OCT of the macula can be used to stage BCD utilizing the following criteria[22].
- Stage 1: drusen-like deposits in the RPE-photoreceptor complex with loss of interdigitation zone and interruption of the ellipsoid layer
- Stage 2: patchy loss of outer retina and RPE
- Stage 3: diffuse RPE atrophy of the posterior pole with extended depth imaging (EDI) showing thinning of the choroid
- Laboratory testing, though not necessary to make the diagnosis of BCD, can be helpful in confirming the diagnosis. Electron microscopy (EM) is confirmatory lysosomal lipid crystalline inclusions are found in lymphocytes and conjunctival fibroblasts.[23] Additionally, inherited retinal disease genetic testing panels that include CYP4V2 gene mutations can be considered.[8]
- Management BCD starts with establishing the diagnosis and referring the patient to a low vision specialist to optimize activities of daily living (ADLs).[3] The baseline examination should include comprehensive ophthalmic exam, retinal imaging, and visual function testing. Rapid decline in vision can be due to CNV which are responsive to anti-vascular endothelial growth factor (VEGF) agents; CME, which responsive to carbonic anhydrase inhibitors, and macular holes which have been successfully treated with vitrectomy with internal limiting membrane (ILM) peel, and gas tamponade.[16][24][25] Genetic testing and counseling for patients early in the disease course (those with preserved photoreceptors) may potentially benefit from emerging gene therapies. At the time of publication there was a single center openly enrolling patients in a phase 1 to explore the efficacy of a subretinal administration of gene replacement therapy by rAAV2/8-hCYP4V2.[26]
- Diagnosis is made when the following triad of symptoms are present: progressive nyctalopia, reduced visual acuity, and visual field loss in the third decade of life.[6][10] Slit-lamp exam can reveal crystals in the perilimbal anterior corneal stroma.[11] On dilated fundus exam (DFE), numerous prominent small, glistening yellow-white crystals are seen scattered throughout the posterior pole extending to the midperiphery. These crystalline deposits may tend to diminish with advanced disease. The yellow-white intraretinal crystals are found at the level of RPE-choriocapillaris complex and lead to RPE pigment clumping and choriocapillaris atrophy. The classic triad of symptoms is also appreciated in diseases on spectrum of retinitis pigmentosa and should be considered in the differential diagnosis.[12] With few ocular diseases sharing common findings of the typical crystalline deposits in the cornea and retina, diagnosis can be made with high degree of certainty from clinical exam alone.[8] There are three stages described by Yuzawa et al[13]:
- Cystinosis is a rare lysosomal storage disease with autosomal recessive inheritance caused by the mutation of the CTNS gene on chromosome 17p13.2 affecting 1 in 100,000-200,000 live births.[27] It affects Europeans more frequently especially in certain regions, such as Brittany in France, likely due to a founder effect. That specific population has an incidence of 1 in 26,000 live births compared to 1 in 325,00 live births in the rest of France. The dysfunctional gene product, cystinosin, impairs cystine transport out of lysosomes leading to its accumulation in the reticuloendothelial cells of the kidneys, bone marrow, pancreas, muscles, brain and eyes.[28][29] For the eyes, the crystal deposition can occur in the conjunctiva, cornea, all retinal layers, and choroid with diffuse RPE degeneration and depigmentation.[30] The exact mechanism of crystalline deposition is unclear; however, it is hypothesized that rampant cellular apoptosis may be a contributing factor.[31]
- Diagnosis is based on age of presentation, location of crystalline deposits, and associated systemic conditions (or lack thereof). The three forms of cystinosis are as follows; in order of incidence:
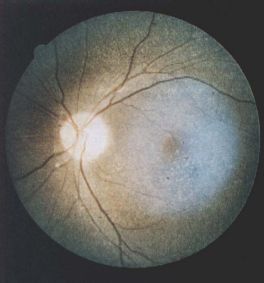 Fine, yellow refractile deposits throughout the retinal layers in cystinosis[32]
Fine, yellow refractile deposits throughout the retinal layers in cystinosis[32]- Infantile nephropathic cystinosis is the most common (95%) and severe presenting within the first year of life with failure to thrive, growth retardation and renal tubular acidosis leading to renal failure within the first decade of life.
- Intermediate nephropathic cystinosis has milder renal impairment, cornea and conjunctival crystals without retinopathy and presents at an older age. They do not develop growth retardation or renal failure.
- Adult ocular cystinosis is the least common and presents with mild photophobia and corneal crystals without nephropathy or retinopathy.[33]
- The first sign of cystinosis is cystine deposits in the cornea and conjunctiva during the first year of life resulting in symptoms of photophobia and blepharospasm.[27][34][33][35][36] The infantile nephropathic cystinosis is the only from having fine, yellow refractile deposits throughout all the retinal layers, RPE and choroid, especially in the periphery of the posterior pole.[3] Diffuse RPE mottling is the most common posterior segment finding that results in a golden-brownish reflex.[37] Pigmentary retinopathy, when present, precedes the appearance of the corneal crystals and can be observed as early as 5 weeks of age.[35][38] With age the retina can display bone spicule pigment migration that resembles retinitis pigmentosa. Progressive retinal degeneration causes decreased visual acuity, constricted visual fields, and decreased cone-rode function.[35] Simultaneously, when photophobia manifests patients with infantile nephropathic cystinosis will have fluid and electrolyte loss, aminoaciduria, glucosuria, phosphaturia, hypercalciuria and hypochloremic acidosis. These patients can present with growth retardation and hypothyroidism.[7]
- Multimodal imaging can play an important role in establishing the diagnosis. SD-OCT can show subfoveal hyporeflectivity and hyperreflective foci in all retinal layers.[39] IVFA angiography will show window defects corresponding to areas of RPE loss and ICGA may show choroidal neovascularization if corneal crystals obscure the view of the fundus.[40] ERG testing shows progressive reduction in photopic and scotopic responses following the evolution of the disease.[41] Reduced visual field and/or nyctalopia can monitored by ERG and visual evoked potentials (VEPs).[42]
- Laboratory testing to measure levels of free non-protein bound cysteine within polymorphonuclear leukocytes is diagnostic.[28] Diagnosis can be delayed due to the rarity of condition and is often recognized when end-stage renal disease is diagnosed.[43] Prenatal diagnostic testing via amniocentesis or chorionic villus sampling is possible for those parents with known risk of having an affected child.[44][45] Many parents elect for diagnostic testing in the early neonatal period with testing for placental cysteine in addition to testing the leukocytes.[27]
- Alternate diagnosis should be considered only in cases with normal cystine concentration in the leukocytes. The main differential diagnosis is Bietti crystalline dystrophy.[41]
- Management of cystinosis centers on cysteamine, an aminothiol, that causes the long-term depletion of lysosomal cystine. If oral cysteamine therapy is initiated prior to symptom development there is benefit in delaying renal failure and subsequent renal transplantation and is also useful in preventing post-transplantation complications.[28] Topical cysteamine drops are effective in removing corneal crystals but have little effect on retinal crystals. If no crystalline retinopathy is present at the time of diagnosis, systemic therapy may prevent retinopathy.[41]
- Diagnosis is based on age of presentation, location of crystalline deposits, and associated systemic conditions (or lack thereof). The three forms of cystinosis are as follows; in order of incidence:
- Primary hyperoxaluria (PH) is a rare group of autosomal recessive inborn errors in hepatic glyoxylate metabolism leading to the overproduction of renally excreted oxalate and glycolate.[46][47] The prevalence is less than 3 in 1,000,000. In 20-50% of cases of severe renal insufficiency, or recurrent renal disease after transplantation occurs before the diagnosis is made. [48] The first signs (recurrent nephrolithiasis, renal colic and infection) appear prior to age of 5 years in 50%, prior to 1 year of age in 10–20% , and by 25 years of age 90 % of the affected patients become symptomatic.[49]
- The three forms are listed below with the associated genetic mutation:
- Diagnosis can be made when in childhood or early adulthood a patient with recurrent calcium oxalate urolithiasis and nephrocalcinosis, resulting in progressive renal damage. The earlier the onset the worse the systemic and visual prognosis. In contrast, the adult onset form has milder urolithiasis and retinopathy.[47][50] Calcium oxalate crystals can be found in the conjunctiva, iris, ciliary body, neurosensory retina, RPE, choroid, and optic nerve.[51][52] If decreased vision is evident optic atrophy is more likely the cause rather than retinal crystal deposits.[50]
- Yellow oxalate crystals are typically found in all retinal layers, though predominantly in the outer plexiform and outer nuclear layers, of the perifoveal or periarterial regions of the macula.[53][50][54] Intraarterial oxalate crystals are associated with secondary hyperoxaluria caused by renal failure from the following causes: methoxyflurane anesthesia, ascorbic acid supplementation, pyridoxine deficiency and ileal resection.[55]
- Crystals are larger in the perifovea, decreasing in size and number moving toward the vascular arcades. Black subretinal ringlets secondary to RPE hyperplasia in response to the crystals can progress to geographic atrophy can have overlying pigment hyperplasia appearing as white fibrous-like tissue overgrowth.[56][30][50][53] The exact mechanism of retinal crystal deposition is unknown.
- The following grading system has been established by Derveaux et al.
- Grade 1: isolated perifoveal oxalate crystals
- Grade 2: macular crystals sparing the fovea with RPE hyperplasia
- Grade 3: RPE hyperplasia, subretinal fibrosis, and/or foveal fibrosis
- Grade 4: macular detachment[53]
- Imaging includes SD-OCT reveals hyperreflective lesions within the dome-shaped RPE elevations and regions of fibrosis and atrophy. FAF shows hyperautofluorescent dots and ring-like areas of hyperautofluorescence surrounding a central hypoautofluorescent area that corresponds to crystalline deposits.[57] EDI-OCT shows the presence of hyperreflective deposits from the retina, subretinal tissue, and choroid.[58] FA reveals a ring of transmitted hyperfluorescence associated with the crystals.[53]
- Diagnosis is based on the presence of oxalate stones associated with nephrocalcinosis and confirmed with the doubling of urinary oxalate excretion in 24-hour urine collection. Genetic testing can identify the specific derangement in oxalate metabolism.[46]
- Differential diagnosis include secondary oxalosis (due to methoxyflurane anaesthesia, ethylene glycol toxicity, xylitol toxicity and chronic renal failure treated by hemodialysis), and other inherited crystalline retinopathies such as Bietti crystalline dystrophy, cystinosis, and Sjögren-Larsson disease.[55]
- Management of hyperoxaluria consists of a diet rich in pyridoxine to decrease urinary calcium oxalate crystallization, optimized hydration, and oral potassium citrate. Once renal failure occurs, dialysis, a combined hepatic and renal transplant should be considered. As oxalate transport is increased the damage to the retina is mitigated.[47] Notably, calcium oxalate deposits are not removed by dialysis. However, regression of the retinal deposits may be noted after successful renal transplantation.[56]
- Sjogren-Larsson Syndrome is an autosomal recessively inherited disorder in fatty acid metabolism caused by the mutation of ALDH3A2 gene on chromosome 17p11.2. This causes fatty aldehyde dehydrogenase (FALDH) deficiency and leads to fatty aldehydes and alcohol accumulation in the skin, eyes, and central nervous system. [59][60][61] Accumulation of the lipid and byproducts of lipid metabolism result in Muller cell damage and dysfunction, neuronal apoptosis, and photoreceptor damage, which is characterized by crystals in the inner nuclear layer. [3] The systemic features are characterized by a triad of congenital ichthyosis, mental retardation, and spastic diplegia.[59][60][61] Though originally described in Sweden the syndrome has been identified in Greece, France, Israel, and Canada. In the US, the Halwi Native Americans of North Carolina were included in the first large case series.[56][59]
- Clinical diagnosis is made with the observation of the triad and is aided by clinical examination noting short stature, speech deficits, and short fingers and toes. Patients often are unable to walk and have seizures as well. Ocular manifestations usually occur by age two; presenting with photophobia, bilateral symmetric vision loss and exam findings including retinal crystals, colobomatous microphthalmia, and congenital cataract. Ecchymosis progressing to ichthyosis can be seen on the upper eyelids.[56][62][63]
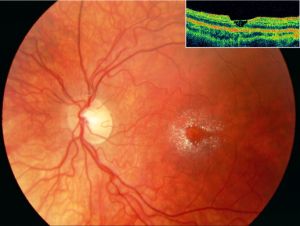 Perimacular crystalline deposits with unremarkable fovea correlating to a cystoid foveal degeneration on OCT in Sjogren-Larsson Syndrome[64]
Perimacular crystalline deposits with unremarkable fovea correlating to a cystoid foveal degeneration on OCT in Sjogren-Larsson Syndrome[64]- Posterior pole exam reveals small, often difficult to detect bilateral, glistening yellow-white dots of varying sizes in the fovea and parafovea that can increase with age.[63] With progression the crystals decreased leading prominent RPE atrophy amount and macular depigmentation. Macular depigmentation likely results from Müller cell dysfunction. The subretinal crystals are likely oxidative byproducts from deranged lipid metabolism and subsequent lipofuscin accumulation correlates to the lack of normal dark yellowish appearance of the central macula.[65][66][67]
- SD-OCT shows the majority of macular crystals in the inner nuclear and outer plexiform layers as well as the ganglion cell and inner plexiform layer.[63][66] Cystic foveal cavitation with foveal atrophy is seen in 67% of patients.[64] Subretinal lipofuscin deposits and ellipsoid loss are also noted.[65] FA shows window defects corresponding to areas of atrophy, crystals without leakage or enlargement of the foveal avascular zone. FAF shows absence of normally reduced foveal autofluorescence due to macular pigment loss.[66] OCTA is notable for decreased retinal capillary density, microvascular dilation, and increased flow voids in the superficial and deep capillary plexuses.[3][63][68]
- Diagnostic testing included genetic testing for biallelic mutation of ALDH3A2 gene or measurement of FALDH activity in cultured fibroblasts.[63][69]
- Differentials include other inherited crystalline retinopathies such as Bietti crystalline dystrophy, cystinosis, and oxalosis.
- Management is supportive and facilitated by a multidisciplinary team including neurology, dermatology, rehabilitation specialists, and geneticist.[70] Fat restriction and supplementation with medium chain triglycerides can also be recommended.[56]
- Clinical diagnosis is made with the observation of the triad and is aided by clinical examination noting short stature, speech deficits, and short fingers and toes. Patients often are unable to walk and have seizures as well. Ocular manifestations usually occur by age two; presenting with photophobia, bilateral symmetric vision loss and exam findings including retinal crystals, colobomatous microphthalmia, and congenital cataract. Ecchymosis progressing to ichthyosis can be seen on the upper eyelids.[56][62][63]
- Kjellin syndrome is a rare autosomal recessive neuro-ophthalmic syndrome characterized by the triad of spastic paraplegia, dementia and macular dystrophy.[71] Consanguineous mating may be present in families of those affected.[72] The syndrome is genetically heterogeneous with two known genes identified in SPG11 and SPG15 whose gene product is spatacsin which is linked to spastic paraplegia with thin corpus callosum.[73] Macular lesions have been noted in heterozygous carriers without evidence of neurologic involvement.[74] Not evident at birth, the syndrome manifests with difficulty in walking and in speech and mental retardation in the first decade of life. By the second decade of life paraplegia becomes the hallmark feature establishing the diagnosis. Progression into the third decade of life leads to dementia.[7] Progressive retinopathy appears between 20 and 35 years of age. Due to devastating neurological effects patients typically have little to visual complaints.[75] The retinal lesions are benign with most patients with mildly decreased vision to 20/30 when lesions are noted in the fovea.[72]
- Diagnosis is based on neurologic signs and is supported by posterior pole findings. Retinal involvement is highly variable and its absence does not rule out Kjellin syndrome.[7] In early stages bilateral symmetric macular or paramacular yellow-white flecks at the level of the RPE aid in diagnosis. These flecks are reminiscent of those seen in Stargardt disease or fundus flavimaculatus, but differ in that the centers are dark or pigmented, encircled by a small halo, and lack pisciform configuration. 50% of patients also have fine whitish dust like lens opacities in the periphery or at the equator.[76][77]
- Multimodal imaging can be helpful in identifying subtle retinal crystals. SD-OCT shows hyperreflective lesions at the level of the RPE. FAF is superior to ophthalmoscopy in identifying flecks that show hyperautofluorescent signal surrounded by dark halo hypoautofluorescent halo.[7][77] Similar to Stargardt disease the FA demonstrates a dark choroid but differs with increased hyperfluorescence of flecks surrounded by a halo of reduced hyperfluorescence. MF-ERG can elicit abnormal responses in the macula.
- Molecular genetic analysis supports the diagnosis.
- Differential diagnosis include Stargardt disease, fundus flavimaculatus, hereditary drusen, fundus albipunctatus, and flecked retina of Kandori.
- There is currently no treatment due to the lack of full characterization of this condition.[76]


Toxic
- Methoxyflurane is, now rarely used, volatile inhalation anesthetic that causes dose-dependent renal tubular damage due to deposition of calcium oxalate crystals resulting in secondary hyperoxalosis. At cumulative doses >16g, calcium oxalate crystals can cause retinopathy during prolonged anesthesia (greater than 4 hours) and in situations of abuse.[56][55][78] Risk factors include mild renal insufficiency and hypertension in addition to exposure.[79][80] Other causes of secondary oxalosis were discussed in the PH section. Deposition of retinal crystals are presumably due to elevated serum levels during prolonged anesthesia. Oxalate crystals have also been noted in the epicardium, bronchus, thyroid, and epididymis.[79] Crystals have been noted in pigment epithelium of the ciliary body and the inner retinal layers.[80]
- Diagnosis is made in the setting of history of methoxyflurane exposure with associated renal dysfunction. Posterior pole exam reveals yellow-white refractile crystals are noted throughout the retina (common intra- or periarterial) and the RPE.[81] FA shows blocking at the level of the RPE corresponding to crystalline deposits.[56]
- Maculopathy is irreversible and cannot be prevented or treated with hemodialysis. Management of renal dysfunction with hemodialysis may be required.[56]
- Tamoxifen citrate is a nonsteroidal estrogen receptor antagonist used as adjuvant therapy in breast cancer treatment.[83] Though the exact mechanism is unclear, refractile yellow-white deposits can be found in a ring-like configuration around the fovea with peripheral deposition being rare. Other ophthalmic manifestations include CME, cystic foveal cavitation, corneal verticillata, cataracts, and optic neuritis. Hallmark features of tamoxifen toxicity include cystic foveal cavitation, crystalline deposits, and macular depigmentation.[4][84][85] Macular edema is present in severe cases.[56] Ocular findings were more evident with cumulative dosing greater than 100g. Current standard dosing, 20 mg daily, has fewer incidences of ocular toxicity; however, even lower doses cause pseudocystic foveal cavitation resembling macular telangiectasia type 2.[86] All of these findings can occur in the absence of visual symptoms or with blurred vision or disturbances in color vision.[4][87] One review cites upwards of 12% retinal toxicity in women taking 20 mg daily.[56] The pathophysiology is thought to be due to its chemical structure which is similar to other medications with retinal toxicity including chloroquine, chlorpromazine, thioridazine, and tilorone. These structurally similar molecules disrupt lysosomal function in the retina and RPE leading potential axonal degeneration and loss of Muller cells. The resulting reactive gliosis of Muller cells ultimately leads to retinal atrophy and cystic foveal cavitation.[88][89]
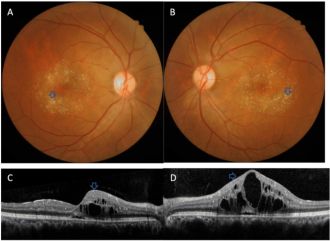 Refractile yellow-white deposits in a ring-like configuration with central CME and hyperreflective deposits in the inner retinal layers in tamoxifen toxicity[82]
Refractile yellow-white deposits in a ring-like configuration with central CME and hyperreflective deposits in the inner retinal layers in tamoxifen toxicity[82]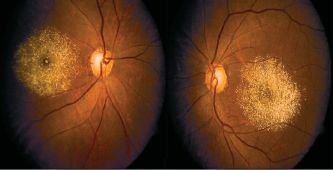 Refractile deposits in the macula in tamoxifen toxicity[90]
Refractile deposits in the macula in tamoxifen toxicity[90]- Diagnosis is based on clinical history and retinal crystals with pigmentary changes and, in severe cases, macular edema.
- Imaging
- SD-OCT shows hyperreflective dots in the nerve fiber layer and inner plexiform layer in addition to cystic foveal cavitations.[4]
- Near infrared reflectance imaging highlights refractile crystals.[91]
- FA can reveal petaloid leakage from CME.[92] Foveal cavitations stain but do not leak.[4][91]
- FAF reveals drastic reduction or absence of normal foveal autofluorescence consistent with pathologies involving macular pigment loss.[4]
- OCTA demonstrates flow voids and anomalies in the deep capillary plexus seen in macular telangiectasia type 2.[93]
- ERG may show decreased photopic and scotopic amplitudes of a- and b-waves; other studies show no retinal toxicity on full-field and focal ERG.[56][94]
- Management
- FA should be obtained in the clinical setting of tamoxifen induced crystalline retinopathy. If there are numerous crystals or macular edema is present the medication should have a dose adjustment or altogether discontinuation. In very early stages retinal crystals are potentially reversible though most cases deposits persist. If maculopathy is not severe visual function and macular edema can improve.[56][45] Cystic foveal cavitations may also persist.[4]
- The CME is responsive to oral acetazolamide, intravitreal anti-VEGF injections, or triamcinolone.[95][96][97]
- Triamcinolone is a synthetic glucocorticoid steroid used as an intravitreal injection for the treatment of cystoid macular edema.[98] Its poor water solubility allows it to have a prolonged effect.[99] The presence of crystals may be explained by failed excretion of steroid crystal particles and subsequent accumulation in the vitreous and eventually the macula due to gravity.
- Diagnosis is made by clinical history of intravitreal triamcinolone injection(s) and posterior pole exam showing large extravascular white to yellow-green iridescent crystals deposits clustered in the fovea or perifovea. Crystals can collect on the retinal surface or in the premacular bursa of the posterior hyaloid.[98][100] Deposition of crystals along the retinal vasculature can simulate frosted-branch angiitis due to their reabsorption by perivascular macrophages.[101]
- OCT imaging shows refractile deposits on the retinal surface; no angiographic abnormality is typically apparent.
- Clinically, no adverse effects on retinal structure or function have been noted in vivo.[98][100] This is potentially due to the vitreous scaffold and internal limiting membrane serving as a barrier to prevent retinal toxicity.[102] Treatment to remove the crystals is not usually necessary.
- Nitrofurantoin is the first line therapy for uncomplicated lower urinary tract infections.[103] Crystalline retinopathy is exceedingly rare and has been reported in one patient who had been on 100mg daily dosing for 19 years (cumulative dose of 690g).
- Diagnosis was established in a patient with mildly decreased vision at 20/30 OU. Posterior pole exam was significant for superficial and deep intraretinal glistening crystalline deposits in a circinate pattern throughout the posterior pole and sparing the periphery
- FA, ERG, and EOG were all normal.[104]
- Reversibility of crystalline deposits is unknown due to the rarity of the complications.
- If encountered in clinical practice the cessation of nitrofurantoin is recommended.[56]
- Canthaxanthin are carotenoid (Vitamin A) derivatives found in fruits, vegetables, and fish. They are red-orange in color and are used in food coloring, dyes, oral tanning agents, and can also be used to treat vitiligo and photosensitivity disorders.[3][105] Retinopathy is variably dose dependent and can occur with a cumulative dose of 19 g in a two-year period.[3][106] Crystals are canthaxanthin-lipoprotein complexes found in areas of spongy degeneration and associated with inner Muller cell atrophy. The complexes are thought to be foci of vascular damage.[107] Risk factors for crystal accumulation include retinal vein occlusion, RPE disruption, and disruption of the inner and outer blood-ocular-barrier occurring in central serous retinopathy. Generally, patients with retinopathy range from asymptomatic with normal vision (common) to having decreased visual acuity, visual field defects, and abnormal ERG findings (low static luminance threshold).[108]
- Diagnosis is based on clinical history and the presence of tiny brilliant golden reflective particles distributed in a donut-shaped configuration in the perifovea and nasal optic disc.[56]
- Patients taking canthaxanthin should be monitored every 6 months for evidence of crystal deposition. Visual field testing is highly sensitive in detecting toxicity as ose dependent decrease in retinal sensitivity has been noted. Cessation of canthaxanthin can reverse retinopathy.[56][111][112] Though it may take up to 20 years for full resolution of crystals there have been no reports of long-term loss of retinal function.[113]
- Lutein & Zeaxanthin are macular xanthophyll pigments concentrated in the fovea. They are the only dietary carotenoids found in the retina.[114] Zeaxanthin predominates centrally with lutein spreading more eccentrically.[115] Low dietary intake of the lutein and zeaxanthin is a risk factor for development of AMD noted in the Carotenoids in Age-Related Eye Disease Study (CAREDS), the Blue Mountain Eye Study, and the Age-Related Eye Disease Study (AREDS).[116] In geographic atrophy secondary to intermediate non-exudative age-related macular degeneration (ARMD) supplement lutein and zeaxanthin prevents vision loss from choroidal neovascularization.[115] Supplementation is well tolerated and without toxic effects.[117] Crystalline retinopathy has been reported twice in the literature. One patient was taking 20mg of lutein daily for 8 years and the other 20mg of zeaxanthin daily for 6 months. Neither case reported vision loss associated with yellow refractile macular crystalline deposits.[114][118]
- The proposed mechanism of inner retinal crystal deposition is the same hypothesized in canthaxanthin toxicity (Muller cell dysfunction). Both processes are due to the ingestion of carotenoids.
- SD-OCT showed crystals in the inner retina.
- FAF revealed diminished intrinsic autofluorescence due to blockage of underlying RPE caused by blockage by macular pigments.
- Differential diagnosis includes canthaxanthin toxicity and West African crystalline retinopathy both of which can be ruled out by comprehensive medical history.[3]
- Zeaxanthin induced crystals resolved with cessation of supplementation in contrast to the lutein induced crystals which persisted despite cessation.[114][118]
- Talc retinopathy is seen in patients with a history of intravenous drug abuse. Talc is filler commonly found in tablets of common medications such as methylphenidate, methadone, and meperidine.[30] Intravenous injection of unfiltered suspension of crushed tablets introduces talc to the pulmonary vascular where the capillary bed allows particles <7μm access to systemic circulation. In the case of inhaled methamphetamine, tiny talc particles enter the systemic circulation via the small capillaries in the nasal mucosa.[119][120][121][122] Symptoms of retinopathy range from asymptomatic crystalline deposits to retinal ischemia from capillary nonperfusion leading to the sequelae of neovascularization.[123] The clinical presentation is similar to sickle cell retinopathy and hemoglobin electrophoresis should aid making the appropriate diagnosis.[1] Clinical history supported by findings of small yellow-white punctate refractile intraarterial crystals scattered throughout the posterior pole.[124] Uncommon manifestations of talc retinopathy include microaneurysms, retinal hemorrhages, cotton wool spots, enlarged arteriolar segments, venous loops, enlarged foveal avascular zone, retinal and choroidal ischemia, and macular fibrosis. These can progress to proliferative abnormalities such as arteriovenous anastomosis, neovascularization of retinal periphery, disc, or iris, vitreous hemorrhage, and tractional retinal detachment.[123][125][126]
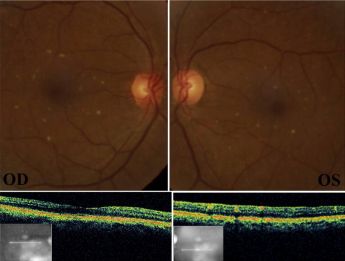 Crystalline talc particles throughout the posterior pole and within vessels[127]
Crystalline talc particles throughout the posterior pole and within vessels[127]- Pathophysiology of talc retinopathy is thought to be due to talc emboli causing a cellular reaction resulting in retinal and choroidal vascular occlusion. Particulates do not necessarily occlude the lumen of precapillary arterioles and capillaries but rather cause focal narrowing. Focal narrowing causes retinal capillary leakage, extensive collateralization of the vascular layer which avoids large retinal infarcts.[128] There are conflicting reports of granulomatous inflammation associated with talc emboli; however, talc is theoretically inert.[128][129]
- Imaging with SD-OCT shows hyperreflective dots of varying size in the nerve fiber layer, ganglion cell layer, inner plexiform and inner nuclear layers.[124][126] Vascular occlusion can cause thinning of the inner retina. Adaptive optics (AO) demonstrate multiple intravascular and extravascular refractile lesions that may not otherwise be detected.[124] The en face OCT image reveals crystals in the superficial vascular network causing shadowing of the deep vascular networks in the central macula. OCT angiography corresponds to the en face imaging demonstrating a normal superficial plexus with focal areas hyporeflectivity in the deep vascular plexus.[130] FA can reveal retinal vascular nonperfusion due to talc embolization, arteriovenous anastomosis, and neovascularization.[1][124] OCT angiography findings include normal superficial plexus, and hyporeflectivity from shadowing in the deep vascular plexus.[130]
- Differential diagnosis of proliferative retinopathy include sickle cell hemoglobinopathies, diabetes, sarcoidosis, systemic lupus erythematosus, leukemic retinopathy, and hyperviscosity retinopathy and should be ruled out with laboratory investigation.[1]
- Proliferative retinopathy should be treated with sectoral PRP.[1]
- Calciphylaxis, also known as calcific uremic arteriolopathy, causing crystalline retinopathy has been reported twice in the literature. It is characterized by calcifications of the medial wall of blood vessels and painful skin ulcers in end-stage renal disease patients undergoing dialysis.[131][132] Vascular occlusion and thrombosis occur secondary to medial wall microcalcification, intimal proliferation, and endovascular fibrosis of small-medium blood vessels, predominantly arteries. The main symptom reported is bilateral decreased visual acuity. Posterior pole exam reveals bilateral crystalline deposits in the macula within the blood vessel walls[131], bilateral pale optic nerves with cotton wool spots and diffuse arteriolar whitening.[132] Intravascular crystals may lead to ischemic manifestations like retinal artery occlusion. Decreased visual acuity is hypothesized to be due to ischemia insults resulting from intravascular crystalline deposits.[131]
- SD-OCT shows multiple dense, deep intraretinal hyperreflective deposits. FAF reveals hyperautoluorescence of the deposits while FAF shows significant delay in choroidal and retinal arterial filling.
- Differential diagnosis is systemic oxalosis in renal failure, which involves crystal deposits in Bruch’s membrane, RPE, and choriocapillaris.[131] One of the case reports had simultaneous ischemic optic neuropathy mimicking giant cell arteritis and crystalline retinopathy with ocular ischemic syndrome separately.
- Empiric intravenous sodium thiosulfate and calcium chelation can improve vision.[132]



Degenerative
- Chronic retinal detachment can lead to fine inner retinal crystals collected in the posterior pole. Crystals are classically seen in peripheral detachment caused by idiopathic retinal dialysis classically seen in the inferotemporal periphery. Punctate crystals are found on the retinal surface no deeper than the internal limiting membrane.[133] Crystals are hypothesized to consist of hemosiderin-laden macrophages, erythrocyte breakdown product secondary to vitreous separation from the internal limiting membrane, or release of photoreceptor outer segment particles.[30][133][134] Earlier investigators identified calcium oxalate and calcium phosphate complexes in the outer retina on histology.[135] The crystals are inert and are not the cause of loss of vision.[136]
- SD-OCT can reveal crystals on the inner surface of the retina. Color fundus photography may help identify subtle crystals.
- Management is centered on treating the retinal dialysis and/or chronic RD with laser retinopexy or cryoretinopexy, scleral buckle, and or pars plana vitrectomy depending on the degree and etiology of the retinal detachment.[3][56]
- Macular telangiectasia type 2 (MacTel Type 2, idiopathic juxtafoveal/macular telangiectasia type 2) is a disorder characterized by retinal capillary telangiectasias in the temporal juxtafoveal region of the macula.[137] Type 1 is a unilateral variant of Coat’s disease seen in youth with 94% male preponderance.[138][139] Type 2 is bilateral, presents more commonly in the middle aged to elderly population with female preponderance and is the only variant with retinal crystals seen in 46% of case.[86] There is high prevalence of diabetes and hypertension found in multiple case series.[140][141][142] There is a genetic component to MacTel Type 2 as it has been identified in monozygotic twins, siblings, and families however no specific gene has been identified yet.[141][143][144][145][146][147][148]
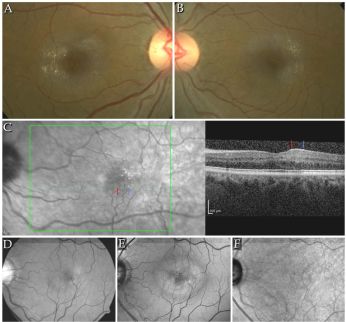 Yellow-golden hyperreflective crystals along telangiectatic vessels with corresponding OCT photos in macular telangiectasia type 2[149]
Yellow-golden hyperreflective crystals along telangiectatic vessels with corresponding OCT photos in macular telangiectasia type 2[149]- The components of the crystals and pathogenesis of the disorder is unknown but may be related to dysfunctional retinoid metabolism in Müller cell affecting cone pigment regeneration and phagocytosis.[86][140] The telangiectasias occur in the superficial and deep retinal capillary plexus causing circinate exudates and CME.[150][151] On fundus exam, small, hyperreflective, white or yellow-golden, superficial retinal crystals are noted in a ring pattern, often temporally along telangiectatic vessels. Progression of disease occurs with reduced retinal transparency, cystoid foveal cavitation, crystal deposition, blunted right angle vessels, pigment plaque formation, macular pigment loss.[152] This leads to subretinal neovascularization that ultimately can cause central vision loss from leakage, hemorrhage, atrophy and scarring.[86][85] Patients can range from asymptomatic to decreased or blurred vision, metamorphopsia, and central scotomas.[30]
- Multimodal imaging may support the diagnosis, primarily detecting telangiectasia manifestations rather than the crystals.[3][56][30] SD-OCT and AO can identify crystals within the nerve fiber layer. Foveal atrophy, cystic foveal cavitation, and foveal and parafoveal retinal thinning are not always evident on SD-OCT.[153][154] Red free and confocal blue light photography can aid in detection of crystals. FA can reveal leakage from the telangiectasias however neither FA or ICG reveal crystalline deposits.[86] OCTA can show dilated vessels in the temporal parafoveal deep capillary plexus early in the disease course and eventual juxtafoveal capillary network with anastomoses and subretinal neovascularization with disease progression.[150][155]
- Differential diagnosis includes other conditions thought to be due Muller cell dysfunction such as Sjogren-Larsson Syndrome and tamoxifen toxicity.[86]
- ENCELTO, an encapsulated, cell-based gene therapy that combines allogeneic retinal pigment epithelial (RPE) cells engineered to secrete recombinant human ciliary neurotrophic factor (hCNTF), is the first therapeutic option for Mactel Type 2. It was shown to have efficacy is slowing the neurodegeneration and preserving photoreceptor structure. ENCELTO is surgically implanted intravitreal device that is sutured to the sclera under aseptic conditions. It was approved for the treatment of adults with MacTel 2 on March 6, 2025.[156]
- Anti-VEGF injections show benefit in the early treatment of small neovascular membranes in MacTel Type 2.
- Refractile/Calcified Drusen Within Geographic Atrophy were first reported by Gass as glistening reflective dot within drusen. Drusen are deposits of end-stage metabolic waste (lipid and protein) from the phototransduction cascade in the RPE that typically accumulate with aging. Deposits are located between Bruch’s membrane and the basement membrane of the RPE.[30][157][158] Calcified drusen, rich in calcium phosphate, in the central macula show depigmentation surrounding the drusen. They are deposited within the four concentric zones of the posterior pole. Small drusen with confluent geographic atrophy (GA) are found in the central macula which represents Zone 1; the innermost zone. Large drusen with confluent GA are found in Zone 2. Zone 3 contains medium drusen without GA. Zone 4, the most outer region, contains small drusen without GA.[159] Vision loss is not usually due to drusen, but rather GA.[160]
- SD-OCT reveals hyperreflective dots on a hyporeflective background without overlying RPE or hyperreflective plaque with RPE atrophy causing increased choroidal transmission.[159][161] FAF demonstrates increased or decreased autofluorescence in areas of calcified drusen and hypoautofluorescence in areas of GA. Near infrared and blue reflectance imaging show hyperreflectivity of drusen.[161]
- Currently, there are no FDA-approved treatments for drusen or GA. AREDS/AREDS2 showed supplementation of specific nutrients to reduce the risk of choroidal neovascular membrane formation and subsequent vision loss.[115]
- Neovascular Age-related Macular Degeneration can cause intraretinal crystalline deposits often associated with prior treatment with various modalities. On exam they are seen as clusters of yellow-white, round-oval crystals of varying size no larger than 30 microns. They are seen with choroidal neovascularization, areas of atrophy or prior macular photocoagulation scar and in areas of prior lipid deposits.[162] Additionally, 5-7% of patients with chronic exudation have intraretinal yellow-grey, refractile, or iridescent patches of organized lipid deposition. They are thought to be subretinal precipitates of cholesterol crystals in an aqueous environment and correspond to a finding called “onion sign.”[163]
- The crystals are easily detected on red-free fundus photography compared to color fundus photography. SD-OCT localizes the crystals to the outer plexiform layer.[163] The “onion sign” can be appreciated as a layered hyperreflective band within a pigment epithelial detachment (PED) correlating with intraretinal and subretinal hyperreflectivity and intraretinal fluid.[163][164]
- No specific treatment is required for both forms of crystal deposits as they do not directly affect visual function.[3]


Idiopathic
- West-African Crystalline Maculopathy (WACM) was described by Sarraf et al. in 2003 as asymmetric bilateral clusters of small refractile yellow-green crystals on the superficial fovea in patients of West African origin. Patient ages range from 40-70 years old without gender predominance. Visual acuity was not affected by crystals.[166] Patients also commonly have associated diabetic retinopathy, CME, sickle cell retinopathy, branch retinal vein occlusion, and familial exudative vitreoretinopathy.[167] The exact composition of the crystals is unclear, though the depositions are hypothesized to be associated with retinal vasculopathies allowing passage through a compromised blood-retinal barrier and possible Müller cell dysfunction.[166][168][169] Foods such as kola nuts, cassava, palm oil, and various leafy green vegetables exclusively to West Africa were proposed as a potential cause; however history of consumption is not consistent.[166][170]
- SD-OCT reveals crystals in either the Henle layer of the fovea or in the innermost layers of the retina.[167][171] FA and electroretinogram are unremarkable.[162]
- No treatment is indicated for WACM.[3][56]
- West African diets which are rich in carotenoids may contribute to crystalline deposition as there are similarities in crystal location and color as seen in canthaxanthin, lutein, and zeaxanthin toxicity.[3][172]
- White Dot Fovea was described in a series of 30 patients in Japan by Yokotsuka et al. in 1997.[173] They report bilateral two to three hundred 5-20 micron white-yellow dots in the fovea in a ring-like pattern. Vision was not affected. The composition of the granules is unknown.
Iatrogenic
- Diamond dusted surgical instruments can leave behind large superficial glistening opacities when used in vitrectomy for epiretinal membrane peel. Originally described in diamond coated silicone cannula; the Tano surgical blade was also reported to shed diamond dust on the retinal surface. The crystals are inert and cause no retinal dysfunction.
References
- ↑ 1.0 1.1 1.2 1.3 1.4 Ruys J, de Smet MD, Van de Sompel W. Bilateral talc maculopathy and fibrovascular proliferation in a drug abuser. Retin Cases Brief Rep. 2010;4(2):123e4
- ↑ Yuzawa M, Mae Y, Matsui M. Bietti’s crystalline retinopathy. Ophthalmic Paediatr Genet. 1986;7(1):9e20
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 3.15 Kovach JL, Isildak H, Sarraf D. Crystalline retinopathy: Unifying pathogenic pathways of disease. Surv Ophthalmol. 2019;64:1-29. doi:10.1016/j.survophthal.2018.08.001
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 Doshi RR, Fortun JA, Kim BT, Dubovy SR, Rosenfeld PJ. Pseudocystic foveal cavitation in tamoxifen retinopathy. Am J Ophthalmol. 2014;157(6):1291e8.e3
- ↑ 5.0 5.1 Mansour AM, Uwaydat SH, Chan CC. Long-term follow-up in Bietti crystalline dystrophy. Eur J Ophthalmol. 2007 Jul-Aug;17(4):680-2. doi: 10.1177/112067210701700434. PMID: 17671952; PMCID: PMC2507722.
- ↑ 6.0 6.1 6.2 6.3 Ng DSC, Lai TYY, Ng TK, Pang CP. Genetics of Bietti Crystalline Dystrophy. Asia-Pacific J Ophthalmol. 2016;5(4):245-252.
- ↑ 7.0 7.1 7.2 7.3 7.4 Puech B, De Laey J-J, Holder GE, eds. Inherited Chorioretinal Dystrophies. Springer Berlin Heidelberg; 2014.
- ↑ 8.0 8.1 8.2 Vargas M, Mitchell A, Yang P, Weleber R. Bietti crystalline dystrophy. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. University of Washington, Seattle; 1993, updated February 7, 2019.
- ↑ Mataftsi A, Zografos L, Millá E, Secrétan M, Munier FL. Bietti’s crystalline corneoretinal dystrophy: a cross-sectional study. Retina. 2004;24(3):416-426.
- ↑ Atmaca LS, Muftuoglu O, Atmaca-Sonmez P. Peripapillary choroidal neovascularization in Bietti crystalline retinopathy. Eye. 2007;21(6):839
- ↑ Bietti G. Ueber familiaeres Vorkommen von ‘Retinitis punctata albescens’ (verbunden mit ‘Dystrophia marginalis cristallinea corneae’), Glitzern des Glaskoerpers und anderen degenerativen Augenveraenderungen. Klin Mbl Augenheilkd. 1937;99:737–57.
- ↑ Zerbib J, Ores R, Querques G, Bouzitou-mfoumou R, Souied EH. Choroidal findings in Bietti’s crystalline dystrophy. Retin Cases Brief Rep. 2014;8(2):130e1
- ↑ Yuzawa M, Mae Y, Matsui M. Bietti’s crystalline retinopathy. Ophthalmic Paediatr Genet. 1986;7(1):9e20
- ↑ 14.0 14.1 Saatci AO, Yaman A, Oner FH, Ergin MH, Cingil G. Indocyanine green angiography in Bietti’s crystalline retinopathy. Can J Ophthalmol. 2002;37(6):346-351.
- ↑ Fong AMY, Koh A, Lee K, Ang CL. Bietti’s crystalline dystrophy in Asians: clinical, angiographic and electrophysiological characteristics. Int Ophthalmol. 2009;29(6):459-470.
- ↑ 16.0 16.1 Fuerst NM, Serrano L, Han G, et al. Detailed functional and structural phenotype of Bietti crystalline dystrophy associated with mutations in CYP4V2 complicated by choroidal neovascularization. Ophthalmic Genetics. 2016;37(4):445-452.
- ↑ Miyake Y. Chapter 2.2. Crystalline Retinopathy (Bietti), Electrodiagnosis of Retinal Diseases. 2nd print. Springer; 2008.
- ↑ Puech B, De Laey J-J, Meunier I. Bietti crystalline corneoretinal dystrophy. In: Puech B, De Laey J-J, Holder GE, eds. Inherited Chorioretinal Dystrophies. Springer Berlin Heidelberg; 2014:355-364.
- ↑ Halford S, Liew G, Mackay DS, et al. Detailed phenotypic and genotypic characterization of bietti crystalline dystrophy. Ophthalmology. 2014;121(6):1174-1184.
- ↑ Kojima H, Otani A, Ogino K, et al. Outer retinal circular structures in patients with Bietti crystalline retinopathy. British Journal of Ophthalmology. 2012;96(3):390-393.
- ↑ Preti RC, Govetto A, Filho RGA, et al. Optical coherence tomography analysis of outer retinal tubulations: sequential evolution and pathophysiological insights. Retina. 2018;38(8):1518-1525.
- ↑ Li Q, Li Y, Zhang X, et al. Utilization of fundus autofluorescence, spectral domain optical coherence tomography, and enhanced depth imaging in the characterization of bietti crystalline dystrophy in different stages. Retina. 2015;35(10):2074-2084.
- ↑ Mansour AM, Uwaydat SH, Chan C-C. Long-term follow-up in Bietti crystalline dystrophy. Eur J Ophthalmol. 2007;17(4):680-682.
- ↑ Broadhead GK, Chang AA. Acetazolamide for cystoid macular oedema in Bietti crystalline retinal dystrophy. Korean J Ophthalmol. 2014;28(2):189e91
- ↑ Nourinia R, Dehghan M, Fekri S. Outcome of macular hole surgery in Bietti crystalline dystrophy. J Ophthalmic Vis Res. 2017;12(3):338.
- ↑ https://clinicaltrials.gov/ct2/show/NCT04722107
- ↑ 27.0 27.1 27.2 Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347(2):111-121.
- ↑ 28.0 28.1 28.2 Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eye drops. Mol Genet Metab. 2000;71(1-2):100-120.
- ↑ Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008;23(6):863-878.
- ↑ 30.0 30.1 30.2 30.3 30.4 30.5 30.6 Nadim F, Walid H, Adib J. The differential diagnosis of crystals in the retina. Int Ophthalmol. 2002;24:113-121.
- ↑ Park MA, Thoene JG. Potential role of apoptosis in development of the cystinotic phenotype. Pediatr Nephrol. 2005;20(4):441-446.
- ↑ Chang BY, George ND. Early blindness due to retinopathy of infantile cystinosis. Eye (Lond). 2000 Oct;14 Pt 5:804-5. doi: 10.1038/eye.2000.218. PMID: 11116719.
- ↑ 33.0 33.1 Gahl WA, Thoene JG, Schneider JA. Cystinosis: a disorder of lysosomal membrane transport. In: Scriver C, Beaudet A, Sly W, et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001.
- ↑ Pinxten A-M, Hua M-T, Simpson J, Hohenfellner K, Levtchenko E, Casteels I. Clinical practice: a proposed standardized ophthalmological assessment for patients with cystinosis. Ophthalmol Ther. 2017;6(1):93-104.
- ↑ 35.0 35.1 35.2 Tsilou E, Zhou M, Gahl W, Sieving PC, Chan C-C. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Survey of Ophthalmology. 2007;52(1):97-105.
- ↑ Shams F, Oladiwura D, Ramaesh K, Livingstone I. Treatment of corneal cystine crystal accumulation in patients with cystinosis. OPTH. Published online October 2014:2077.
- ↑ Dufier JL, Dhermy P, Gubler MC, et al. Ocular changes in long- term evolution of infantile cystinosis. Ophthalmic Paediatr Genet. 1987;8:131–7.
- ↑ Wong VG, Lietman PS, Seegmiller JE. Alterations of pigment epithelium in cystinosis. Archives of Ophthalmology. 1967;77(3):361-369.
- ↑ Adhi M, Duker JS. Optical coherence tomography – current and future applications: Current Opinion in Ophthalmology. 2013;24(3):213-221.
- ↑ Tsilou E, Csaky K, Rubin BI, Gahl W, Kaiser-Kupfer M. Retinal visualization in an eye with corneal crystals using indocyanine green videoangiography. Am J Ophthalmol. 2002;134(1):123e5
- ↑ 41.0 41.1 41.2 Puech B, De Laey J-J, Meunier I. Cystinosis. In: Puech B, De Laey J-J, Holder GE, eds. Inherited Chorioretinal Dystrophies. Springer Berlin Heidelberg; 2014:355-364.
- ↑ Biswas S, Gaviria M, Malheiro L, Marques JP, Giordano V, Liang H. Latest clinical approaches in the ocular management of cystinosis: a review of current practice and opinion from the ophthalmology cystinosis forum. Ophthalmol Ther. 2018;7(2):307-322.
- ↑ Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrology Dialysis Transplantation. 2014;29(suppl 4):iv87-iv94.
- ↑ Schneider JA, Verroust FM, Kroll WA, et al. Prenatal diagnosis of cystinosis. N Engl J Med 1974;290:878-882
- ↑ 45.0 45.1 Smith ML, Pellett OL, Cass MM, et al. Prenatal diagnosis of cystinosis utilizing chorionic villus sampling. Prenat Diagn 1987;7:23-26
- ↑ 46.0 46.1 46.2 Monico CG, Rossetti S, Belostotsky R, et al. Primary hyperoxaluria type iii gene hoga1 (Formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. CJASN. 2011; 6(9): 2289-2295.
- ↑ 47.0 47.1 47.2 47.3 Cochat P, Rumsby G. Primary hyperoxaluria. Ingelfinger JR, ed. N Engl J Med. 2013;369(7):649-658.
- ↑ Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290-296.
- ↑ Gagnadoux MF, Lacaille F, Niaulet P, Revillon Y, Jouvet P, Jan D, Guest G, Charbit M. Broyer M Long term results of liver-kidney transplantation in children with primary hyperoxaluria. Pediatr Nephrol. 2001;16:946–50.
- ↑ 50.0 50.1 50.2 50.3 Small KW. Ocular findings in primary hyperoxaluria. Arch Ophthalmol. 1990;108(1):89.
- ↑ Sakamoto T, Maeda K, Sueishi K, Inomata H, Onoyama K. Ocular histopathologic findings in a 46-year-old man with primary hyperoxaluria. Arch Ophthalmol. 1991;109(3):384e7
- ↑ Brini A Oxalose. In: Offret G, Dhermy P, Brini A, Bec P, editors. Anatomie Pathologique de L’Oeil et de ses Annexes. Paris: Masson; 1974, p. 204.
- ↑ 53.0 53.1 53.2 53.3 Derveaux T, Delbeke P, Walraedt S, et al. Detailed clinical phenotyping of oxalate maculopathy in primary hyperoxaluria type 1 and review of the literature. Retina. 2016;36(11):2227-2235.
- ↑ Roth BM, Yuan A, Ehlers JP. Retinal and choroidal findings in oxalate retinopathy using edi-oct. Ophthalmic Surg Lasers Imaging. 2012;43(6):S142-S144.
- ↑ 55.0 55.1 55.2 Wells CG. Retinal oxalosis: a clinicopathologic report. Arch Ophthalmol. 1989;107(11):1638.
- ↑ 56.00 56.01 56.02 56.03 56.04 56.05 56.06 56.07 56.08 56.09 56.10 56.11 56.12 56.13 56.14 56.15 56.16 56.17 56.18 Drenser K, Sarraf D, Jain A, Small KW. Crystalline Retinopathies. Surv Ophthalmol. 2006;51(6):535-549. doi:10.1016/j.survophthal.2006.08.006
- ↑ Querques G, Bouzitou-Mfoumou R, Soubrane G, Souied EH. Spectral-domain optical coherence tomography visualisation of retinal oxalosis in primary hyperoxaluria. Eye Lond Engl. 2010;24(5):941e3
- ↑ Ozisik GG, Asena L, Bulam B, Gungor SG. Enhanced depth imaging optical coherence tomography features in a young case of primary hyperoxaluria Type 1. Retin Cases Brief Rep. 2015;9(1):92e4
- ↑ 59.0 59.1 59.2 Sjogren T, Larsson T: Oligophrenia in combination with congenital ichthyosis and spastic disorders: a clinical and genetic study. Acta Psychiat Neurol Scand 32:1--112, 1957
- ↑ 60.0 60.1 Rogers GR, Markova NG, De Laurenzi V, et al: Genomic organization and expression of the human fatty aldehyde dehydrogenase gene (FALDH). Genomics 39:127--35, 1997
- ↑ 61.0 61.1 Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opinion on Orphan Drugs. 2016;4(4):395-406.
- ↑ Willemsen MA, Cruysberg JR, Rotteveel JJ, Aandekerk AL, Van Domburg PH, Deutman AF. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjogren-Larsson syndrome. Am J Ophthalmol. 2000;130(6):782e9
- ↑ 63.0 63.1 63.2 63.3 63.4 Jack LS, Benson C, Sadiq MA, Rizzo WB, Margalit E. Segmentation of retinal layers in Sjogren-larsson syndrome. Ophthalmology. 2015;122(8):1730e2
- ↑ 64.0 64.1 Fuijkschot J, Cruysberg JRM, Willemsen MAAP, Keunen JEE, Theelen T. Subclinical changes in the juvenile crystalline macular dystrophy in Sjogren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115(5):870e5
- ↑ 65.0 65.1 Nanda T, Kovach JL. Ophthalmic findings in late stage sjogren–larsson syndrome. RETINAL Cases & Brief Reports. 2019;13(3):251-254
- ↑ 66.0 66.1 66.2 van der Veen RLP, Fuijkschot J, Willemsen MAAP, Cruysberg JRM, Berendschot TTJM, Theelen T. Patients with Sjogren-Larsson syndrome lack macular pigment. Ophthalmology. 2010;117(5):966e71
- ↑ Reichenbach A, Bringmann A. New functions of Muller cells. Glia. 2013;61(5):651e78.
- ↑ Jean-Franc ̧ ois E, Low JY, Gonzales CR, Sarraf D. Sjogren- larsson syndrome and crystalline maculopathy associated with a novel mutation. Arch Ophthalmol Chic Ill 1960. 2007;125(11):1582e3
- ↑ Rizzo WB, Craft DA: Sjo ̈gren-Larsson syndrome. Deficient activity of the fatty aldehyde dehydrogenase component of fatty alcohol:NADþ oxidoreductase in cultured fibroblasts. J Clin Invest 88:1643--8, 1991
- ↑ Bindu PS. Sjogren-larsson syndrome: mechanisms and management. Appl Clin Genet. 2020;13:13-24.
- ↑ Kjellin KG, Stibler H. Protein patterns of cerebrospinal fluid in hereditary ataxias and hereditary spastic paraplegia. Journal of the Neurological Sciences. 1975;25(1):65-74.
- ↑ 72.0 72.1 Farmer SG, Longstreth WT, Kalina RE, et al: Fleck retina in Kjellin’s syndrome. Am J Ophthalmol 99:45--50, 1985
- ↑ Stevanin G, Santorelli FM, Azzedine H, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007;39(3):366-372.
- ↑ Seidel K, De Vos R, Derksen L, et al. Widespread thalamic and cerebellar degeneration in a patient with a complicated hereditary spastic paraplegia (Hsp). Annals of Anatomy - Anatomischer Anzeiger. 2009;191(2):203-211.
- ↑ Ferriby D, Stojkovic T, De Seze J, et al. Kjellin syndrome [in French]. Rev Neurol (Paris) 2001;157:80–3.
- ↑ 76.0 76.1 Frisch IB, Haag P, Steffen H, Weber BHF, Holz FG. Kjellin’s syndrome. Ophthalmology. 2002;109(8):1484-1491.
- ↑ 77.0 77.1 Sachdev A, Proudlock FA, Abbott R, Gottlob I. Kjellin syndrome: First case with retinal changes in carriers. Neurology. 2005;65(7):1110-1110.
- ↑ Mazze RI. Fluorinated anaesthetic nephrotoxicity: an update. Can Anaesth Soc J. 1984;31(S3):S16-S22.
- ↑ 79.0 79.1 Bullock JD, Albert DM. Flecked retina. Appearance secondary to oxalate crystals from methoxyflurane anesthesia. Arch Ophthalmol Chic Ill 1960. 1975;93(1):26e31
- ↑ 80.0 80.1 Bullock JD, Albert DM, Skinner HC, Miller WH, Galla JH. Calcium oxalate retinopathy associated with generalized oxalosis: x-ray diffraction and electron microscopic studies of crystal deposits. Invest Ophthalmol. 1974;13(4):256-265.
- ↑ Novak MA, Roth AS, Levine MR. Calcium oxalate retinopathy associated with methoxyflurane abuse. Retina Phila Pa. 1988;8(4):230e6
- ↑ Li C, Xiao J, Zou H, Yang B, Luo L. The response of anti-VEGF therapy and tamoxifen withdrawal of tamoxifen-induced cystoid macular edema in the same patient. BMC Ophthalmol. 2021 May 7;21(1):201. doi: 10.1186/s12886-021-01953-z. PMID: 33962570; PMCID: PMC8106132.
- ↑ Kaiser-Kupfer MI, Lippman ME. Tamoxifen retinopathy. Cancer Treat Rep. 1978;62(3):315e20
- ↑ Bourla DH, Sarraf D, Schwartz SD. Peripheral retinopathy and maculopathy in high-dose tamoxifen therapy. Am J Ophthalmol. 2007;144(1):126e8
- ↑ 85.0 85.1 Salomão SR, Watanabe SES, Berezovsky A, Motono M. Multifocal electroretinography, color discrimination and ocular toxicity in tamoxifen use. Curr Eye Res. 2007;32(4):345e52
- ↑ 86.0 86.1 86.2 86.3 86.4 86.5 Sallo FB, Leung I, Chung M, et al. Retinal crystals in type 2 idiopathic macular telangiectasia. Ophthalmology. 2011;118(12):2461e7
- ↑ Lazzaroni F, Scorolli L, Pizzoleo CF, et al. Tamoxifen retinopathy: does it really exist? Graefe’s Archive for Clinical and Experimental Ophthalmology. 1998;236(9):669-673.
- ↑ Dyer MA, Cepko CL. Control of Muller glial cell proliferation and activation following retinal injury. Nat Neurosci. 2000;3(9):873e80
- ↑ Gualino V, Cohen SY, Delyfer M-N, Sahel J-A, Gaudric A. Optical coherence tomography findings in tamoxifen retinopathy. Am J Ophthalmol. 2005;140(4):757e8
- ↑ Nair SN, Anantharaman G, Gopalakrishnan M, Vyas J. Spectral domain optical coherence tomography findings in tamoxifen retinopathy--a case report. Retin Cases Brief Rep. 2013 Spring;7(2):128-30. doi: 10.1097/ICB.0b013e31825956f1. PMID: 25390802.
- ↑ 91.0 91.1 Neuville J, Yevseyenkov V. Spectral domain oct imaging techniques in tamoxifen retinopathy. Optometry and Vision Science. 2015;92(2):e55-e59.
- ↑ Kaiser-Kupfer MI, Kupfer C, Rodrigues MM. 1. Tamoxifen retinopathy. Ophthalmology. 1981;88(1):89-93.
- ↑ Todorich B, Yonekawa Y, Thanos A, Randhawa S. Oct angiography findings in tamoxifen maculopathy. Ophthalmology Retina. 2017;1(5):450-452.
- ↑ Watanabe SES, Berezovsky A, Motono M, et al. Retinal function in patients treated with tamoxifen. Doc Ophthalmol. 2010;120(2):137-143.
- ↑ Jeng KW, Wheatley HM. Intravitreal triamcinolone acetonide treatment of tamoxifen maculopathy with associated cystoid macular edema. RETINAL Cases & Brief Reports. 2015;9(1):64-66.
- ↑ Zafeiropoulos P, Nanos P, Tsigkoulis E, Stefaniotou M. Bilateral macular edema in a patient treated with tamoxifen: a case report and review of the literature. Case Rep Ophthalmol. 2014;5(3):451-454.
- ↑ Rahimy E, Sarraf D. Bevacizumab therapy for tamoxifen-induced crystalline retinopathy and severe cystoid macular edema. Arch Ophthalmol. 2012;130(7):931.
- ↑ 98.0 98.1 98.2 Sarraf D, Vyas N, Jain A, et al. Triamcinolone-associated crystalline maculopathy. Arch Ophthalmol. 2010;128(6):685-690.
- ↑ Gopal L, Sharma T. Use of intravitreal injection of triamcinolone acetonide in the treatment of age-related macular degeneration. Indian J Ophthalmol. 2007;55(6):431-435.
- ↑ 100.0 100.1 Shaikh S, Zarifa R, Kester E. Persistence of triamcinolone crystals after intravitreal injection: Benign crystalline hyaloidopathy. Indian J Ophthalmol. 2013;61(4):182.
- ↑ García-Campos JM, García-Basterra I, Kamal-Salah R, Baquero-Aranda I. Retinal pseudoangiitis after intravitreal triamcinolone. BMJ Case Rep. 2015;2015.
- ↑ Spitzer MS, Mlynczak T, Schultheiss M, et al. Preservative-free triamcinolone acetonide injectable suspension versus “traditional” triamcinolone preparations: impact of aggregate size on retinal biocompatibility. Retina. 2011;31(10):2050-2057.
- ↑ Huttner A, Verhaegh EM, Harbarth S, Muller AE, Theuretzbacher U, Mouton JW. Nitrofurantoin revisited: a systematic review and meta-analysis of controlled trials. J Antimicrob Chemother. 2015;70(9):2456-2464.
- ↑ Ibanez HE. Crystalline retinopathy associated with long-term nitrofurantoin therapy. Arch Ophthalmol. 1994;112(3):304.
- ↑ Harnois C, Cortin P, Samson J, Boudreault G, Malenfant M, Rousseau A. Static perimetry in canthaxanthin maculopathy. Arch Ophthalmol Chic Ill 1960. 1988;106(1):58e60
- ↑ Boudreault G, Cortin P, Corriveau LA, Rousseau AP, Tardif Y, Malenfant M. Canthaxanthine retinopathy: 1. Clinical study in 51 consumers. Can J Ophthalmol J Can Ophtalmol. 1983;18(7):325e8
- ↑ Sujak A. Interactions between canthaxanthin and lipid membranes–possible mechanisms of canthaxanthin toxicity. Cell Mol Biol Lett. 2009;14(3):395e410
- ↑ Fraunfelder FW. Ocular side effects from herbal medicines and nutritional supplements. Am J Ophthalmol. 2004;138(4):639e47
- ↑ Chan A, Ko TH, Duker JS. Ultrahigh-resolution optical coherence tomography of canthaxanthine retinal crystals. Ophthalmic Surg Lasers Imaging. 2006;37(2):138e9
- ↑ Espaillat A, Aiello LP, Arrigg PG, Villalobos R, Silver PM, Cavicchi RW. Canthaxanthine retinopathy. Arch Ophthalmol Chic Ill 1960. 1999;117(3):412e3
- ↑ Harnois C, Samson J, Malenfant M, Rousseau A. Canthaxanthin retinopathy. Anatomic and functional reversibility. Arch Ophthalmol. 1989;107(4):538-540.
- ↑ Leyon H, Ros AM, Nyberg S, Algvere P. Reversibility of canthaxanthin deposits within the retina. Acta Ophthalmol (Copenh). 1990;68(5):607-611.
- ↑ Hueber A, Rosentreter A, Severin M. Canthaxanthin retinopathy: long-term observations. Ophthalmic Res. 2011;46(2):103e6
- ↑ 114.0 114.1 114.2 Choi RY, Gorusupudi A, Wegner K, Sharifzadeh M, Gellermann W, Bernstein PS. Macular pigment distribution responses to high-dose zeaxanthin supplementation in patients with macular telangiectasia type 2. Retina Phila Pa. 2017;37(12):2238e47
- ↑ 115.0 115.1 115.2 Age-Related Eye Disease Study 2 (AREDS2) Research Group, Chew EY, Clemons TE, et al. Secondary analyses of the effects of lutein/zeaxanthin on age-related macular degeneration progression: AREDS2 report No. 3. JAMA Ophthalmol. 2014;132(2):142e9
- ↑ Weigert G, Kaya S, Pemp B, et al. Effects of lutein supplementation on macular pigment optical density and visual acuity in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(11):8174.
- ↑ van de Kraats J, Kanis MJ, Genders SW, van Norren D. Lutein and zeaxanthin measured separately in the living human retina with fundus reflectometry. Invest Ophthalmol Vis Sci. 2008;49(12):5568e73
- ↑ 118.0 118.1 Choi RY, Chortkoff SC, Gorusupudi A, Bernstein PS. Crystalline maculopathy associated with high-dose lutein supplementation. JAMA Ophthalmol. 2016;134(12):1445e8
- ↑ Kumar RL, Kaiser PK, Lee MS. Crystalline Retinopathy from Nasal Ingestion of Methamphetamine. Retina. 2005;26(7):823-824.
- ↑ AtLee WE. Talc and cornstarch emboli in eyes of drug abusers. JAMA. 1972;219(1):49e51
- ↑ Brucker AJ. Disk and peripheral retinal neovascularization secondary to talc and cornstarch emboli. Am J Ophthalmol. 1979;88(5):864e7
- ↑ Schatz H, Drake M. Self-injected retinal emboli. Ophthalmology. 1979;86(3):468e83
- ↑ 123.0 123.1 Jampol LM, Setogawa T, Rednam KR, Tso MO. Talc retinopathy in primates. A model of ischemic retinopathy: I. Clinical studies. Arch Ophthalmol Chic Ill 1960. 1981;99(7):1273e80
- ↑ 124.0 124.1 124.2 124.3 Soliman MK, Sarwar S, Hanout M, et al. High-resolution adaptive optics findings in talc retinopathy. Int J Retina Vitr. 2015;1:10
- ↑ Sharma MC, Ho AC. Macular fibrosis associated with talc retinopathy. Am J Ophthalmol. 1999;128(4):517e9
- ↑ 126.0 126.1 Tse DT, Ober RR. Talc retinopathy. Am J Ophthalmol. 1980;90(5):624e40
- ↑ Shah VA, Cassell M, Poulose A, Sabates NR. Talc retinopathy. Ophthalmology. 2008 Apr;115(4):755-755.e2. doi: 10.1016/j.ophtha.2007.10.043. PMID: 18387416.
- ↑ 128.0 128.1 Kaga N, Tso MO, Jampol LM. Talc retinopathy in primates: a model of ischemic retinopathy. III. An electron microscopic study. Arch Ophthalmol Chic Ill 1960. 1982;100(10):1649e57
- ↑ Michelson JB, Whitcher JP, Wilson S, O’Connor GR. Possible foreign body granuloma of the retina associated with intravenous cocaine addiction. American Journal of Ophthalmology. 1979;87(3):278-280.
- ↑ 130.0 130.1 Bagheri N, Shahlaee A, Sridhar J, Ho AC. En face optical coherence tomography and angiography of talc retinopathy. Acta Ophthalmol (Copenh). 2016;94(1):103e4
- ↑ 131.0 131.1 131.2 131.3 Naysan J, Dansingani KK, Balaratnasingam C, et al. Crystalline retinopathy and retinal vasculopathy in calcific uremic arteriolopathy(Calciphylaxis). RETINAL Cases & Brief Reports. 2018;12(4):331-335.
- ↑ 132.0 132.1 132.2 Cherayil NR, Scoles D, Moran AM, Elder DE, Tamhankar MA. Gazing into the crystal ball: calciphylaxis causing striking retinal vascular calcification, ocular ischemic syndrome, crystalline retinopathy, and ischemic optic neuropathy. Journal of Neuro-Ophthalmology. 2020;Publish Ahead of Print.
- ↑ 133.0 133.1 Ahmed I, McDonald HR, Schatz H, et al. Crystalline retinopathy associated with chronic retinal detachment. Arch Ophthalmol. 1998;116(11):1449-1453.
- ↑ Habib MS, Byrne S, McCarthy JH, Steel DHW. Refractile superficial retinal crystals and chronic retinal detachment: case report. BMC Ophthalmol. 2006;6:3
- ↑ Cogan DG, Kuwabara T, Silbert J, Kern H, McMURRAY V, Hurlbut C. Calcium oxalate and calcium phosphate crystals in detached retinas. AMA Arch Ophthalmol. 1958;60(3):366e71
- ↑ Alassane S, Binquet C, Arnould L, et al. Spatial distribution of macular pigment in an elderly french population: the montrachet study. Invest Ophthalmol Vis Sci. 2016;57(10):4469-4475.
- ↑ Gass JD, Oyakawa RT. Idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol Chic Ill 1960. 1982;100(5):769e80
- ↑ Abujamra S, Bonanomi MT, Cresta FB, Machado CG, Pimentel SL, Caramelli CB. Idiopathic juxtafoveolar retinal telangiectasis: clinical pattern in 19 cases. Ophthalmologica. 2000;214(6):406-411.
- ↑ Abujamra S, Bonanomi MT, Cresta FB, Machado CG, Pimentel SL, Caramelli CB. Idiopathic juxtafoveolar retinal telangiectasis: clinical pattern in 19 cases. Ophthalmologica. 2000;214(6):406-411.
- ↑ 140.0 140.1 Clemons TE, Gillies MC, Chew EY, et al. Baseline characteristics of participants in the natural history study of macular telangiectasia (MacTel) MacTel Project Report No. 2. Ophthalmic Epidemiol 2010;17:66–73.
- ↑ 141.0 141.1 Chew EY, Murphy RP, Newsome DA, et al. Parafoveal telangiectasis and diabetic retinopathy. Arch Ophthalmol 1986;104:71–5.
- ↑ Barbazetto IA, Room M, Yannuzzi NA, et al. ATM gene variants in patients with idiopathic perifoveal telangiectasia. Invest Ophthalmol Vis Sci 2008;49:3806–11.
- ↑ Gillies MC, Zhu M, Chew EY, et al. Familial asymptomatic macular telangiectasia type 2. Ophthalmology 2009;116:2422–9.
- ↑ Hannan SR, Madhusudhana KC, Rennie C, et al. Idiopathic juxtafoveolar retinal telangiectasis in monozygotic twins. Br J Ophthalmol 2007;91:1729–30.
- ↑ Menchini U, Virgili G, Bandello F, et al. Bilateral juxtafoveolar telangiectasis in monozygotic twins. Am J Ophthalmol 2000;129:401–3.
- ↑ Hutton WL, Snyder WB, Fuller D, et al. Focal parafoveal retinal telangiectasis. Arch Ophthalmol 1978;96:1362–7.
- ↑ Isaacs TW, McAllister IL. Familial idiopathic juxtafoveolar retinal telangiectasis. Eye 1996;10(5):639–42.
- ↑ Oh KT, Park DW. Bilateral juxtafoveal telangiectasis in a family. Retina 1999;19:246–7.
- ↑ Sallo FB, Leung I, Chung M, Wolf-Schnurrbusch UE, Dubra A, Williams DR, Clemons T, Pauleikhoff D, Bird AC, Peto T; MacTel Study Group. Retinal crystals in type 2 idiopathic macular telangiectasia. Ophthalmology. 2011 Dec;118(12):2461-7. doi: 10.1016/j.ophtha.2011.05.022. Epub 2011 Aug 12. PMID: 21839520; PMCID: PMC3433242.
- ↑ 150.0 150.1 Roisman L, Rosenfeld PJ. Optical coherence tomography angiography of macular telangiectasia type 2. Dev Ophthalmol. 2016;56:146e58
- ↑ Spaide RF, Klancnik JM, Cooney MJ, et al. Volume- rendering optical coherence tomography angiography of macular telangiectasia type 2. Ophthalmology. 2015;122(11):2261e9
- ↑ Yannuzzi LA, Bardal AMC, Freund KB, Chen K-J, Eandi CM, Blodi B. Idiopathic macular telangiectasia. 2006. Retina Phila Pa. 2012;32(Suppl 1):450e60
- ↑ Charbel Issa P, Kupitz EH, Heeren TFC, Holz FG. Treatment for macular telangiectasia type 2. Dev Ophthalmol. 2016;55:189e95
- ↑ Toto L, Di Antonio L, Mastropasqua R, et al. Multimodal imaging of macular telangiectasia type 2: focus on vascular changes using optical coherence tomography angiography. Invest Ophthalmol Vis Sci. 2016;57(9):268e76
- ↑ Tan GS, Kuehlewein L, Sadda SR, Sarraf D, Schwartz SD. Subretinal neovascularization in macular telangiectasia type 2: optical coherence tomographic angiography and treatment response. Retin Cases Brief Rep. 2015;9(4):286-289.
- ↑ Hoy SM. Revakinagene Taroretcel: First Approval. Mol Diagn Ther. 2025 Jul;29(4):553-561. doi: 10.1007/s40291-025-00787-5. Epub 2025 Jun 20. Erratum in: Mol Diagn Ther. 2025 Sep;29(5):705. doi: 10.1007/s40291-025-00802-9. PMID: 40540142; PMCID: PMC12343658.
- ↑ Gass JDM. Drusen and disciform macular detachment and degeneration. Archives of Ophthalmology. 1973;90(3):206-217.
- ↑ Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48(3):257-293.
- ↑ 159.0 159.1 Suzuki M, Curcio CA, Mullins RF, Spaide RF. Refractile drusen: clinical imaging and candidate histology. Retina. 2015;35(5):859-865.
- ↑ Bressler NM, Bressler SB, Fine SL. Age-related macular degeneration. Surv Ophthalmol. 1988;32(6):375-413.
- ↑ 161.0 161.1 Oishi A, Thiele S, Nadal J, et al. Prevalence, natural course, and prognostic role of refractile drusen in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2017;58(4):2198-2206.
- ↑ 162.0 162.1 Lima LH, Freund KB, Klancnik JM, Spaide RF. Intraretinal crystalline deposits in neovascular age-related macular degeneration. Retina. 2010;30(4):542-547.
- ↑ 163.0 163.1 163.2 Pang CE, Messinger JD, Zanzottera EC, Freund KB, Curcio CA. The onion sign in neovascular age-related macular degeneration represents cholesterol crystals. Ophthalmology. 2015;122(11):2316-2326.
- ↑ Mukkamala SK, Costa RA, Fung A, Sarraf D, Gallego-Pinazo R, Freund KB. Optical coherence tomographic imaging of sub-retinal pigment epithelium lipid. Arch Ophthalmol. 2012;130(12):1547-1553.
- ↑ Weng CY, Morales JF, Gupta I. West African Crystalline Maculopathy in a Nigerian Woman. Ophthalmology. 2018 Mar;125(3):390. doi: 10.1016/j.ophtha.2017.11.022. PMID: 29458828.
- ↑ 166.0 166.1 166.2 Sarraf D, Ceron O, Rasheed K, Drenser KA, Casey R. West African crystalline maculopathy. Arch Ophthalmol. 2003;121(3):338-342.
- ↑ 167.0 167.1 Rajak SN, Mohamed MD, Pelosini L. Further insight into West African crystalline maculopathy. Arch Ophthalmol. 2009;127(7):863-868.
- ↑ Browning DJ. West African crystalline maculopathy. Ophthalmology. 2004;111(5):921-925.
- ↑ Or C, Kirker AW, Forooghian F. Uveitic crystalline maculopathy. J Ophthalmic Inflamm Infect. 2015;5:5.
- ↑ Kung JS, Leng T. West african crystalline maculopathy in sickle cell retinopathy. Case Rep Ophthalmol Med. 2015;2015:910713.
- ↑ Baker PS, Ho AC, Spirn MJ. Optical coherence tomography of west african crystalline maculopathy. Retin Cases Brief Rep. 2009;3(1):31-32.
- ↑ Smith G, Dueker S, Clifford A, Grivetti L. Carotenoid values of selected plant foods common to Southern Burkina Faso, West Africa. Ecol of Food & Nutrition. 1996;35(1):43-58.
- ↑ 173.0 173.1 Yokotsuka K, Kishi S, Shimizu K. White dot fovea. Am J Ophthalmol. 1997;123(1):76-83.
- ↑ Witkin AJ, London NJS, Wender JD, Fu A, Garg SJ, Regillo CD. Spectral-domain optical coherence tomography of white dot fovea. Arch Ophthalmol. 2012;130(12):1603.
- ↑ Lewis JM, Park I, Ohji M, Saito Y, Tano Y. Diamond-dusted silicone cannula for epiretinal membrane separation during vitreous surgery. Am J Ophthalmol. 1997;124(4):552-554.